


About Article
Elucidating impacts of deleterious Missense mutations on the structure and function of beta-glucuronidase: Investigating the molecular basis of pathogenesis of MPSVII
Abstract
Single-nucleotide polymorphisms (SNPs) are among the most prevalent forms of genetic variation and are frequently associated with human diseases. In this study, we performed a comprehensive in silico analysis of non-synonymous SNPs (nsSNPs) in the GUSB gene, which encodes the lysosomal enzyme β-glucuronidase, a key regulator of glycosaminoglycan degradation. A total of 449 reported mutations were systematically evaluated using sequence- and structure-based predictive algorithms. Among these, 15 variants were identified as deleterious and structurally destabilizing. Subsequent pathogenicity assessment using SNPs&GO, MutPred, and PhD-SNP further narrowed these to eight high-confidence pathogenic mutations. Solubility and aggregation propensity analysis using the SODA tool revealed that 37.5% of these pathogenic variants showed increased aggregation or reduced solubility, suggesting a potential mechanism contributing to disease progression. Detailed structural investigation indicated that the observed destabilization likely arises from alterations in interatomic non-covalent interactions, ultimately compromising protein stability and function. Overall, this study provides a systematic and integrative characterization of pathogenic nsSNPs in the GUSB gene. The findings enhance our understanding of the structural and functional consequences of these variants and offer a foundation for future experimental validation and the development of targeted therapeutic strategies.
Keywords
1. Introduction
β-glucuronidase (GUSB) is a crucial enzyme widely used in molecular biology and genetic engineering projects. (Yang et al., 2017). The housekeeping enzyme GUSB plays an active role in proteoglycan synthesis in lysosomes and is expressed in most tissues. It contributes significantly to the gradual decline in dermatan and keratan sulfates by enhancing the disintegration of GAGs during the fifth cycle. GUSB promotes the dispersion of active or inert compounds from glucuronides, thereby modifying the manner in which prodrugs respond and operate (Naz et al., 2013).
GUSB deficiency leads to mucopolysaccharidosis type VII, resulting in brain lysosomal storage (Kong et al., 2022). Humans have been shown to exhibit 11 distinct types (MPS I to IIIA, B, C, and IV to IX), which are divided according to the deficient enzyme (Hytonen et al., 2012). There are seven different types of Mucopolysaccharidosis (MPS), which are categorized based on defects in a certain number of the eight specific lysosomal metabolic enzymes (Khan et al., 2017). Mutations in the IDUA gene on chromosome 4p16 cause the first kind of MPS, which is caused by a deficiency of the enzyme a-l-iduronidase. A-l-iduronidase deficiency results in the accumulation of GAGs, such as dermatan and heparan sulphates (DS and HS), in many human tissues, leading to severe organ malfunction (Nagpal et al., 2022).
MPS II is linked to lysosomal dysfunction due to a deficiency of the enzyme iduronate 2-sulphatase, which degrades heparan sulphate and dermatan sulphate (DS). A rapidly developing neurodegenerative lysosomal storage disease is MPSIII. It is caused by biallelic variants in a gene encoding an enzyme in the digestive tract that degrades heparan sulfate. Neuronal inflammation and significant activation of astrocytes and microglia are hallmarks of MPS III, a neurological disease (Seker Yilmaz et al., 2021). MPS IVA, additionally referred to as Morquio syndrome type A, constitutes one of the most significant lysosomal illnesses. The lysosomal hydrolase N-acetylglucosamine-6-sulfate sulfatase (GALNS) is absent in this autosomal-recessive genetic disorder (Sawamoto et al., 2020). There is nothing abnormal regarding the nervous system's functions, but common features include elevated blood and urine potassium (K+) levels, a petite stature, odontoid hypoplasia, pectus carinatum, kyphoscoliosis, genu valgum, joint laxity, and corneal clouding (Tomatsu et al., 2011).
Many lysosomal storage disorders, including mucopolysaccharidosis type VII (MPS VII), commonly referred to as Sly syndrome, are frequently linked to these mutations (?). The integrity and activity of β-glucuronidase are dramatically affected by missense, frameshift, and nonsense mutations in the GusB gene, according to recent investigations (?). GusB Gene encodes a 651-amino-acid-long homotetrameric protein with three distinctive domains in every monomer (Florindo et al., 2018). One such relationship involves the lysosomal-associated membrane protein 2 (LAMP2), which supports the stability and appropriate transit of β-glucuronidase inside lysosomes (Staudt et al., 2016). Furthermore, heparan sulfate and chondroitin sulfate are broken down by β-glucuronidase, which works in tandem with other enzymes such as arylsulfatase B and heparanase (Naz et al., 2013).
According to (Urayama et al., 2004), it is imperative that GUSB reach its intended intracellular location via this targeting mechanism. The lysosomal route of GAG breakdown is the main pathway in which GUSB is involved. An essential stage in the breakdown of GAGs is the cleavage of the β-D-glucuronic acid residues by GUSB. GAG buildup is caused by a deficit in β-glucuronidase activity and results in lysosomal storage disorders (Hytonen et al., 2012). GAGs (glycosaminoglycans) are broken down by many lysosomal enzymes in an intricate procedure. The remaining molecules of β-D-glucuronic acid from GAGs such as heparan sulfate, dermatan sulfate, and chondroitin sulfate are particularly susceptible to hydrolysis by GUSB (Awolade et al., 2020).
GUSB utilizes its N-terminus domain to identify and adhere to a GAG chain, its substrate. The catalytic function of the enzyme in question depends on the initial binding (Urayama et al., 2004). The site that catalyzes the breakdown of β-D-glucuronic acid residues is located in the catalytic region of GUSB (Hytonen et al., 2012). After splitting, the active region of the enzyme discharges the resulting molecules, which are shorter oligosaccharides, enabling β-Glucuronidase to interact with a different substrate (Staudt et al., 2016). Supporting autophagy and lysosomal activity requires β-glucuronidase. It supports the prevention of cellular malfunction by facilitating the breakdown of autophagic organelles. The amino acids associated with LAMP2 maintain its continued existence within lysosomes and ensure a steady supply of functional GUSB for the degradation of lysosomal proteins (Staudt et al., 2016).
GUSB is involved in the processes of extracellular matrix reorganization and cell-mediated cascade modulation, in addition to lysosomal breakdown, by controlling the accessibility of functional glycosaminoglycan fragments (Wan et al., 2020). By interacting with receptors located on the cell surface, like integrins and growth factor receptors, GAG fragments can encourage cytoskeleton remodeling and improve cell motility (Casale & Crane, 2024).
The residues of breakdown may change the extracellular matrix’s mechanical features and the substance, which may affect how cells engage with their surroundings (Wang et al., 2017). Pattern recognition receptors (PRRs) on immune cell populations identify particular glycosaminoglycan components as danger-associated molecular patterns (DAMPs), which then, in turn, cause inflammatory reactions (Garantziotis & Savani, 2022). About carcinoma, the breakdown components of glycosaminoglycan might affect the activity of the cancerous cells and the stromal cells that envelop them, therefore facilitating the growth, blood vessel development, and territorial expansion of the tumor (Hua et al., 2022). Prospective treatment options for recovering β-glucuronidase function in MPS VII patients include gene therapy and enzyme replacement therapy (ERT) (Grubb et al., 2010). Furthermore, the use of small-molecule chaperones to regulate or increase the efficiency of mutant β-glucuronidase has already been investigated (Doherty et al., 2023).
Single-nucleotide polymorphisms (SNPs), particularly non-synonymous SNPs (nsSNPs), are major contributors to genetic variability and are frequently implicated in inherited metabolic disorders. Despite numerous reported variants, a systematic approach to distinguish pathogenic mutations from benign polymorphisms remains limited. Given the increasing volume of genomic data generated through high-throughput sequencing, there is a critical need to prioritize functionally significant variants using reliable approaches. Identifying deleterious nsSNPs and understanding their structural and functional consequences can provide valuable insights into disease mechanisms, genotype–phenotype correlations, and molecular instability associated with protein dysfunction. Therefore, this study was undertaken to comprehensively analyze nsSNPs in the GUSB gene using integrative sequence- and structure-based bioinformatics tools. By predicting pathogenicity, assessing structural destabilization, and evaluating aggregation propensity, the work aims to bridge the gap between genetic variation data and mechanistic understanding. Ultimately, such insights may facilitate improved diagnostic interpretation, biomarker identification, and the development of targeted therapeutic interventions.
2. Materials and methods
2.1. Data retrieval and analysis
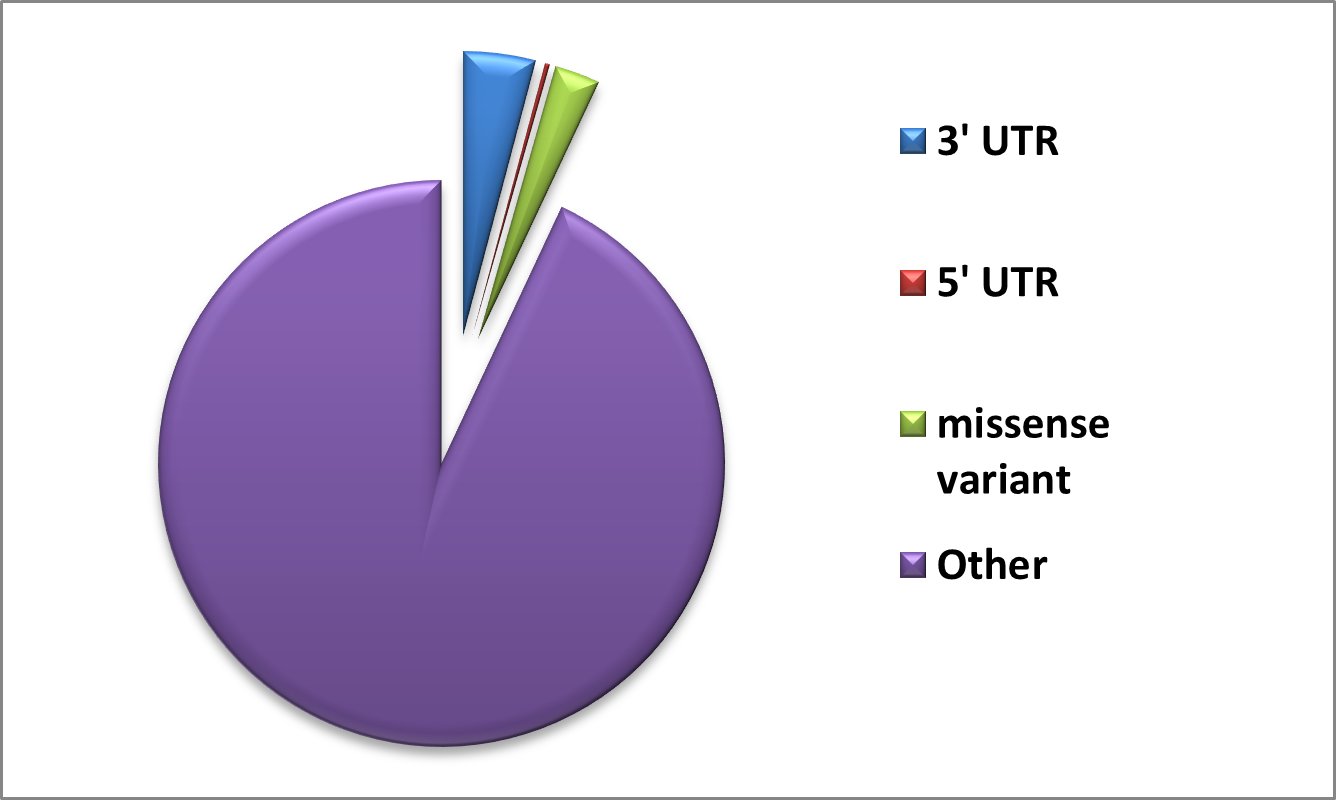
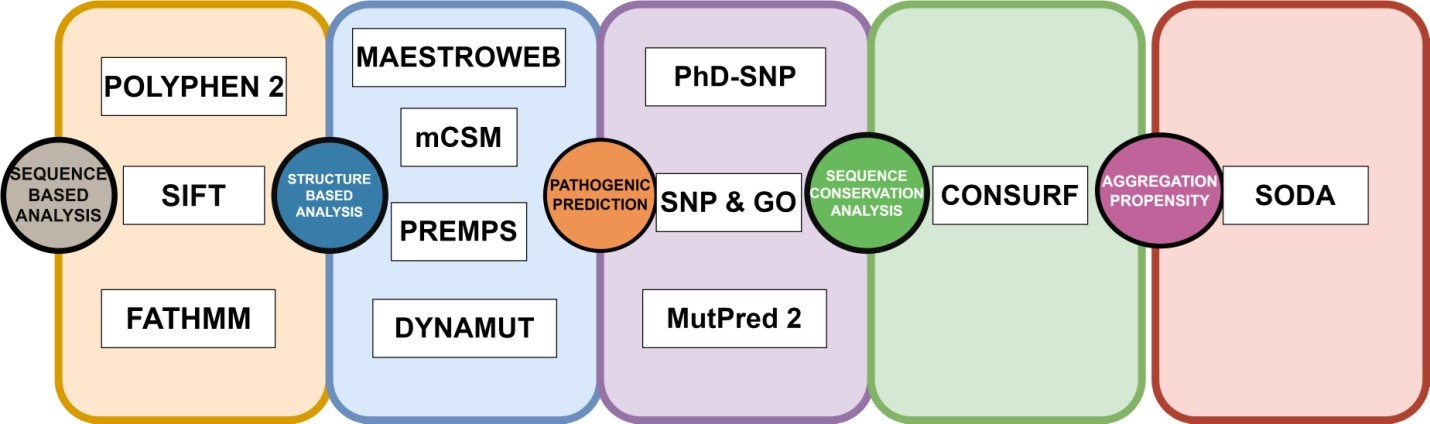
The GUSB sequence was extracted in FASTA format from the UniProt repository (UniProt ID: P08236). A collection of non-synonymous SNPs was generated using information obtained from the PubMed literature search and databases such as dbSNP (Sherry et al., 2001), HGMD (Stenson et al., 2009), ClinVar (Landrum et al., 2014), and Ensembl (Hubbard et al., 2002). The list was cleared to eliminate redundant nsSNPs. The protein data bank provided the crystal structure of the human GUSB gene. Out of the total Data, 1305 Missense variants along with 3’UTR and 5’ UTR were obtained from dbSNP and Ensembl, and are shown in Figure. We used a comprehensive computational approach to anticipate harmful mutations in the GUSB at both structural and functional levels, as illustrated in Figure.
2.2. Sorting Intolerant from Tolerant
Sorting Intolerant from Tolerant (SIFT) was used to predict how possible amino acid alterations may affect protein function. SIFT is expanded to include predictions for frame-shifting indels. Human genetic research has extensively used SIFT to assess amino acid substitutions (e.g., cancer, Mendelian disorders, and viral infections). Studies of human diseases and other study subjects are not the only areas in which SIFT is useful. The consequences of missense mutations on model animals such as rats, dogs, and Arabidopsis, as well as agricultural plants, have been studied using SIFT (Sim et al., 2012). If the SIFT score is less than or equal to 0.05, then the mutation is not tolerable (Ng & Henikoff, 2003). The SIFT method was utilized to forecast the impact of these nsSNPs on the protein.
2.3. Polymorphism Phenotyping v2
Polymorphism Phenotyping v2 (PolyPhen-2), a program and Web server accessible, estimates the potential effects of amino acid alterations on the stability and activity of human proteins. SNPs are functionally annotated; coding SNPs are mapped to gene transcripts; protein sequence annotations are extracted; and conservation profiles are constructed. The likelihood that the missense mutation would cause harm is then calculated using a combination of all these characteristics (Adzhubei et al., 2013). In addition to estimating the likelihood that a mutation is harmful when it is not, PolyPhen-2 also provides predictions of false-positive and true-positive rates. PolyPhen-2 computes the naive Bayes posterior probability that a particular mutation is harmful. The qualitative assessment of a mutation determines whether it is benign, perhaps harmful, or likely harmful (Adzhubei et al., 2010).
2.4. Mutation Assessor
Mutation Assessor (http://mutationassessor.org/r3/) is a sequence-based tool that predicts the functional impact of an nsSNPs on a protein. This server predicts the functional effects of missense polymorphisms and amino acid modifications observed in cancer-causing mutations in proteins. Depending on the affected amino acids and the degree of evolutionary preservation in protein homologs, the functional impact is evaluated (?). A sizable collection of polymorphic and disease-associated (OMIM) variants—60,000 in total has been used to verify the technique.
2.6. Mutation Cutoff Scanning Matrix
A computer method called Mutation Cutoff Scanning Matrix (mCSM) is used to predict how mutations may affect small-molecule binding, protein-protein interactions, protein-nucleic acid interactions, and protein stability. mCSM is an innovative tool for assessing non-synonymous mutations that predicts destabilizing mutations using a graph-based method. If a mutation results in a mCSM score (ΔΔ G) less than 0, it affects protein structure (Choudhury et al., 2021). It assists in forecasting the impact of mutations on drug binding, hence contributing to the development of more potent medications. It helps understand how changes in protein interactions and stabilization lead to conditions caused by mutations. Directs the creation of proteins for use in industry and medicine that have the appropriate stability and interaction characteristics (Pires et al., 2016).
2.7. MAESTRO
The free energy change on protein unfolding is estimated using MAESTRO (https://pbwww.che.sbg.ac.at/maestro/web), a multi-agent stability prediction tool. It determines how a point mutation affects the protein's stability by calculating the difference in free energy (Δ G) between the mutant and wild-type proteins. The stability of the protein is changed by a mutation if its score is less than zero (Laimer et al., 2015). Mutations may be limited to certain amino acid classes, exposed or buried residues, and user-specified areas, contingent upon the purpose of the research (Laimer et al., 2015).
2.8. PREMPS
PremPS calculates changes in the unfolding Gibbs free energy to assess the effects of single mutations on protein stability. This approach requires a protein's three-dimensional structure (Chen et al., 2020). The PREMPS software determines the mutation's corresponding change in free energy (ΔΔ G). PREMPS forecasts the impact of the mutation on protein stability based on the calculated ΔΔ G. Generally, a destabilizing mutation is indicated by a high ΔΔ G value, whereas a stabilizing mutation is suggested by a negative ΔΔ G (Chen et al., 2020).
2.9. DynaMut
DynaMut forecasts the effects of mutations on protein stability by considering MD simulation outcomes. The protein structure is stabilized or destabilized by a mutation based on the calculation of metrics such as changes in free energy (ΔΔ G). This prediction is essential for evaluating how mutations in biological systems will affect functionality (Rodrigues et al., 2018). PhD-SNP is a tool designed to help understand the genetic basis of illnesses. It analyzes SNPs by assessing their possible negative consequences using a variety of structural and evolutionary factors. A prediction score that indicates the probability of an SNP being deleterious is produced by PhD-SNP. A greater score denotes a higher likelihood that the SNP would result in functional alterations or increased susceptibility to illness, with a score range from 0 to 1 (Capriotti et al., 2006).
2.10. SNP & GO
A bioinformatics technique called SNP & GO (http://snps-and-go.biocomp.unibo.it/snps-and-go/) integrates data from both Gene Ontology (GO) and SNP annotations to forecast the functional effects of SNPs. This tool facilitates understanding of genomic data in relation to disease and phenotype by illuminating the potential effects of genetic variants on protein function and biological mechanisms. The algorithms used here categorize SNPs as neutral or deleterious (likely to impair protein function) based on the attributes that were retrieved. SNP & GO produces prediction scores determined by the machine learning model that show how likely it is that an SNP will have negative effects. Higher scores indicate a greater likelihood of functional impact, which helps researchers choose SNPs for further study (Capriotti et al., 2013).
2.11. MutPred2
MutPred2 was created to predict how missense mutations in human proteins might affect protein function. It evaluates how mutations influence protein structure, function, and interactions by combining diverse features and machine learning methods, delivering significant insights into their potential roles in disease processes (Choudhury et al., 2021).
2.12. Solubility based on Disorder and Aggregation
A technique to determine the aggregation, disorder, helix, and strand propensities resulting from mutations is called Solubility based on Disorder and Aggregation (SODA) (http://protein.bio.unipd.it/soda/). The PDB format structure file or the protein sequence may be entered into this program. SODA uses Fells, ESpritz-NMR, PASTA 2.0, and other resources to forecast mutations of many types, including insertions, deletions, substitutions, and duplications. Based on how differently the WT and mutant proteins are soluble, SODA assigns a final score (Paladin et al., 2017).
2.13. Consurf Analysis
Consurf (https://consurf.tau.ac.il/) is a bioinformatics tool designed to find areas in protein sequences that have been conserved across time. It uses evolutionary links among various species and sequence homology to predict the structural and functional significance of amino acid residues. Using multiple sequence alignment, the ConSurf tool was used to assess residue conservation at a particular location (Ashkenazy et al., 2016).
3. Results and Discussion
A thorough investigation was carried out to obtain missense mutations of the GUSB gene using several well-known genetic databases, including Ensembl (http://www.ensembl.org/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), HGMD (http://www.hgmd.cf.ac.uk), and dbSNP (http://www.ncbi.nlm.nih.gov/snp), HGMD (http://www.hgmd.cf.ac.uk), and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/). A total of 1305 missense mutations (nsSNPs) were found after an extensive search. Since certain nsSNPs may not have been indexed in these databases, a thorough PubMed literature review was conducted to ensure a more comprehensive collection. The primary objective of the present study was to conduct a comprehensive evaluation of all reported missense mutations in the GUSB gene. To understand their inheritance patterns and potential biological consequences, each variant was initially assessed at the sequence level. This approach enables a deeper understanding of how individual mutations may alter protein function and contribute to disease pathogenesis (?).
To ensure strong, high-confidence identification of deleterious variants, we adopted an integrative strategy combining sequence- and structure-based computational analyses. The sequence-based assessment was first performed using well-established tools, including SIFT, PolyPhen-2, Mutation Assessor (http://mutationassessor.org/r3/), and FATHMM. Because reliance on a single predictive algorithm can lead to false positives or false negatives, multiple tools were employed to improve prediction accuracy and minimize bias. SIFT classifies nsSNPs as tolerated or deleterious based on evolutionary conservation and amino acid physicochemical properties. Variants with lower tolerance index scores are predicted to have a greater functional impact on the protein (Ng & Henikoff, 2003). PolyPhen-2 evaluates the possible impact of amino acid substitutions on protein structure and function using comparative and structural features (Adzhubei et al., 2010; Adzhubei et al., 2013). Mutation Assessor predicts the functional consequences of mutations by categorizing them into different impact levels (e.g., low, medium, or high) based on evolutionary conservation of affected residues (?). FATHMM further enhances prediction reliability by integrating sequence conservation with hidden Markov models to assess the likelihood of pathogenicity (Shihab et al., 2013).
Subsequently, structure-based tools such as MAESTROweb (https://pbwww.che.sbg.ac.at/maestro/web), PremPS, mCSM, and DynaMut were employed to evaluate the effects of mutations on protein stability, conformational dynamics, and intermolecular interactions. This combined framework provides a more reliable and comprehensive understanding of the structural and functional implications of nsSNPs in the GUSB gene (Chen et al., 2020; Pires et al., 2016; Rodrigues et al., 2018; Laimer et al., 2015).
3.1. Sequence-based predictions
Mutation Assessor categorizes variants according to their predicted functional impact. Moderate-impact mutations are expected to slightly alter protein stability or activity, whereas high-impact mutations are predicted to severely impair protein function, potentially leading to loss of activity or altered biological behavior. Similarly, FATHMM classifies variants into two principal categories based on their anticipated effects: tolerated and damaging. Tolerated variants are predicted to have little or no functional consequence and are likely benign or neutral. In contrast, damaging variants are expected to significantly disrupt protein stability, activity, or molecular interactions, thereby increasing their likelihood of being pathogenic (Shihab et al., 2015). These predictive frameworks enable researchers to prioritize high-risk variants for experimental validation and clinical investigation, particularly those identified as deleterious for disease-association and functional studies.
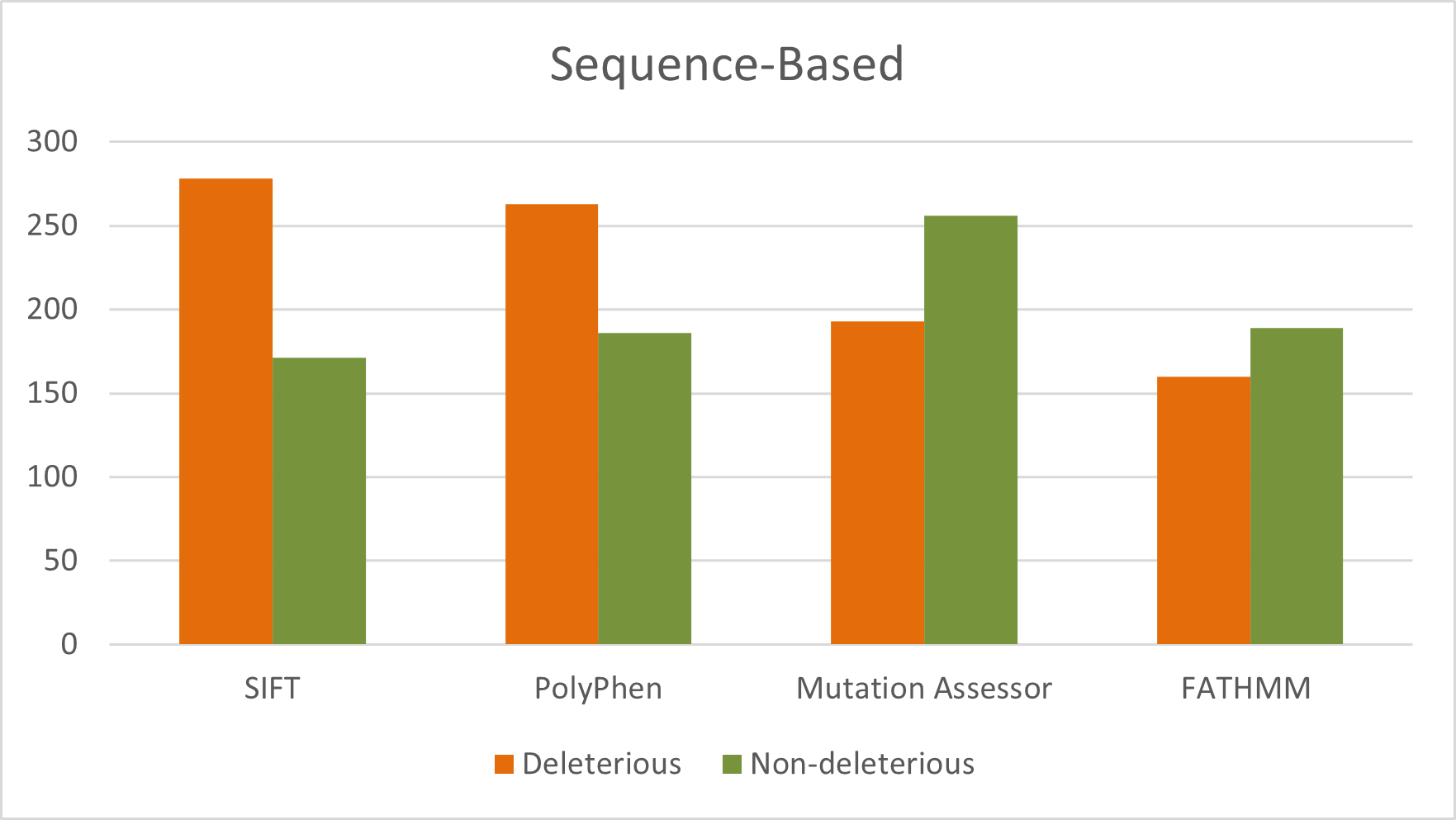
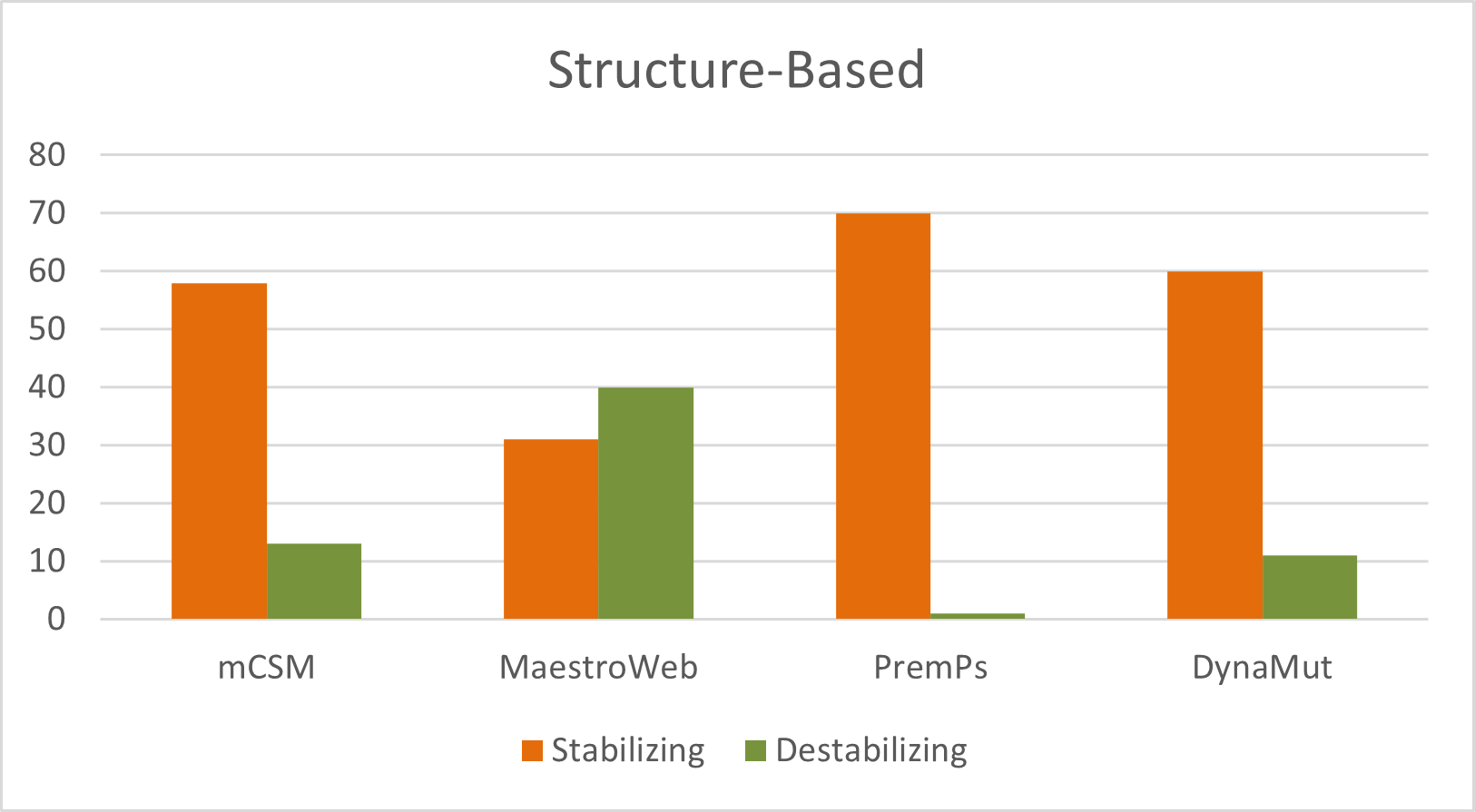
A total of 449 nsSNPs in the human GUSB gene were analyzed. Sequence-based predictions identified 277, 262, 160, and 192 deleterious mutations using SIFT, PolyPhen-2, FATHMM, and Mutation Assessor, respectively. Structural stability analysis revealed 13, 40, 1, and 11 destabilizing mutations as predicted by mCSM, MAESTROweb, PremPS, and DynaMut, respectively (Figure). Overall, this comprehensive multi-tool strategy strengthens confidence in variant prioritization and provides a reliable foundation for subsequent functional validation and disease-correlation studies (Supplementary Table S1).
3.2. Structure-based prediction
In addition to sequence-based analyses, four structure-based stability prediction tools DynaMut, PremPS, mCSM, and MAESTROweb were employed in this study. These programs estimate changes in folding free energy (ΔΔ G) using atomic coordinates derived from the wild-type protein’s PDB structure. Most of these tools integrate biophysical principles with machine-learning or neural-network–based approaches to evaluate the impact of amino acid substitutions on protein stability and conformational dynamics. To enhance prediction reliability and minimize false positives, only those variants consistently identified as deleterious by all four sequence-based tools and all four structure-based methods were selected for further analysis. This stringent integrative screening approach resulted in 15 high-confidence mutations predicted to be both deleterious and structurally destabilizing (Figure).
mCSM employs graph-based signatures to predict the impact of amino acid substitutions on protein stability by quantifying changes in folding free energy (ΔΔ G). Using this approach, mCSM identified 13 destabilizing mutations. MAESTROweb predicted 40 mutations as destabilizing, utilizing a combination of structural descriptors, statistical potentials, and energetic parameters to estimate the effects of mutations on protein stability and functionality. PremPS (Predictions of Protein Stability Changes upon Mutation) identified one destabilizing mutation. This tool integrates structural environmental features with probabilistic energy functions to assess the effects of single-point mutations on protein stability. DynaMut detected 11 destabilizing mutations by combining graph-based signatures with normal-mode analysis to assess their effects on both protein stability and conformational dynamics (Supplementary Table S2).
3.3. Identification of pathogenic nsSNPs
To further investigate the disease associations of the prioritized nsSNPs in the human GUSB gene, we employed three well-established pathogenicity prediction tools: PhD-SNP, SNPs&GO, and MutPred. These computational platforms classify variants as benign or pathogenic based on probability scores derived from sequence features, evolutionary conservation, and functional annotations, thereby enabling reliable assessment of disease association. From our integrative sequence- and structure-based analyses, 15 nsSNPs were identified as high-confidence deleterious and destabilizing variants. These shortlisted mutations were subsequently subjected to pathogenicity evaluation using the three prediction tools.
PhD-SNP classified 12 out of the 15 variants (80%) as pathogenic, indicating a strong likelihood of disease association. Similarly, SNPs&GO predicted 10 of the 15 variants (66.66%) to be disease-related, further supporting the potential clinical relevance of these mutations (Table). To enhance confidence and minimize false-positive predictions, we compared results from PhD-SNP and SNPs&GO. Eight variants were consistently identified as pathogenic by both tools, namely: L214P, L566P, N135D, P108L, R26L, T180P, Y388C, and Y399C. These eight mutations were therefore prioritized as the most probable disease-causing variants and selected for subsequent detailed analysis.
3.4. Analysis of evolutionarily conserved residues
Analysis of amino acid conservation within a protein structure provides critical insight into the functional and structural importance of specific residues and reflects the evolutionary constraints that shape them. Highly conserved residues are often essential for maintaining structural integrity, catalytic activity, or molecular interactions. In general, the likelihood of mutation tolerance is inversely related to the degree of conservation; residues that are strongly conserved across species are less likely to accommodate substitutions without functional consequences.
To evaluate evolutionary conservation in the human GUSB protein, we performed a ConSurf analysis (Figure). The results indicated that residues within the regions 360–370, 380–390, 415–425, and 566–571 exhibit a high degree of conservation compared to other segments of the protein. These conserved regions likely play critical roles in maintaining structural stability and functional competence. Furthermore, ConSurf classification revealed that several of these highly conserved residues are buried within the protein core, suggesting a structural role in maintaining stability, while others are exposed on the protein surface, potentially contributing to functional interactions. These findings reinforce the biological significance of mutations occurring within conserved regions of the GUSB protein.
3.5. Analysis of aggregation propensity
Amino acid solubility plays a critical role in maintaining proper protein folding and function (Balch et al., 2008). Previous studies have demonstrated that reduced protein solubility can promote misfolding and aggregation, which are central mechanisms underlying several neurodegenerative and aggregation-related disorders (Knowles et al., 2014). Insoluble protein regions tend to cluster, leading to pathological aggregates associated with diseases such as Parkinson’s disease, Alzheimer’s disease, and amyloidosis (Thal et al., 2015).
SNPs are the most common form of genetic variation and are widely associated with diverse human diseases. Comprehensive analysis of these variants provides valuable insight into the molecular mechanisms underlying disease progression and may facilitate the development of targeted therapeutic strategies. In this study, we focused on nsSNPs in the GUSB gene, a critical lysosomal hydrolase responsible for glycosaminoglycan degradation.
To evaluate the effects of prioritized variants on the solubility of the GUSB protein, we used the SODA tool. SODA provides a comprehensive assessment by estimating mutation-induced changes in aggregation propensity, intrinsic disorder, helix propensity, and strand propensity. This integrative analysis enables a deeper understanding of how specific amino acid substitutions may influence protein folding behavior and aggregation potential (Choudhury et al., 2021).
From our earlier pathogenicity assessment, eight nsSNPs were identified as disease-associated variants (Table 2). These eight mutations were subsequently analyzed using SODA to determine their impact on protein solubility. Among them, three variants were predicted to reduce protein solubility. Decreased solubility is typically associated with increased aggregation propensity, which may contribute to disease pathogenesis by forming aberrant protein assemblies. These findings suggest that these three variants could potentially promote aggregation-mediated dysfunction of the GUSB protein.
In contrast, the remaining five nsSNPs were predicted to enhance protein solubility. Increased solubility generally reflects a reduced tendency to aggregate and may help maintain a more stable, functionally competent protein conformation. Such mutations may therefore have a comparatively lower risk of inducing aggregation-related pathogenic effects. The analysis demonstrated that three of the eight pathogenic mutations (37.5%) are predicted to decrease protein solubility and increase aggregation tendency. This observation is particularly significant, as protein aggregation is a well-established contributor to several pathological conditions, including neurodegenerative and aggregation-related disorders. In contrast, the remaining variants were predicted to maintain or enhance solubility, suggesting comparatively lower aggregation risk.
Overall, the SODA analysis expands our understanding of how specific GUSB variants may influence protein solubility and aggregation behavior. This comprehensive computational analysis highlights a subset of GUSB nsSNPs with strong evidence of deleterious, destabilizing, and pathogenic effects. By integrating assessments of stability, pathogenicity, and solubility, the study provides a detailed molecular perspective on how specific variants may impair β-glucuronidase function and contribute to disease pathogenesis.
To further evaluate the pathogenic potential of the prioritized variants, we employed three robust and widely validated bioinformatics tools, SNPs&GO, MutPred, and PhD-SNP, recognized for their reliability in predicting the disease association of SNPs. These analyses consistently identified eight mutations: L214P, L566P, N135D, P108L, R26L, T180P, Y388C, and Y399C as highly deleterious, suggesting significant implications for protein stability, functional integrity, and disease susceptibility.
To gain additional structural and evolutionary insight, we performed conservation analysis using the ConSurf server, which evaluates the evolutionary conservation of amino acid residues within a protein sequence (Ashkenazy et al., 2016). The results revealed that two of the eight pathogenic mutations, L566P and Y388C, are located within highly conserved regions of the protein. Such strong evolutionary conservation typically reflects critical structural or functional importance. Therefore, substitutions at these positions are likely to exert substantial effects on protein stability and activity, further supporting their potential pathogenic role.
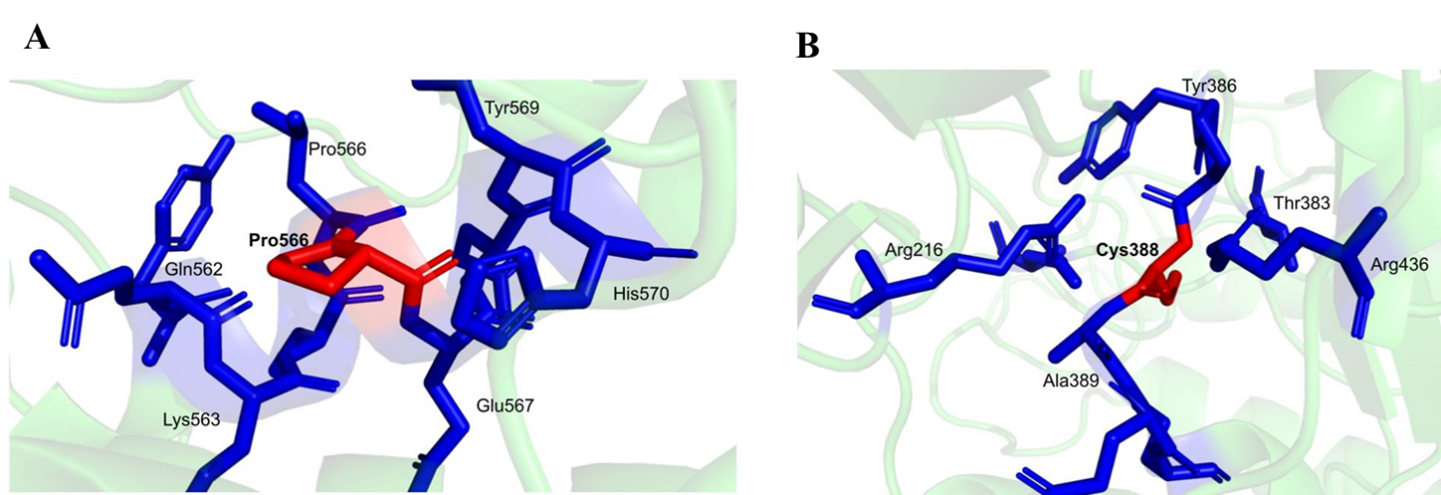
3.6. Analyzing Structures of Specific Mutations
To gain deeper insight into the molecular consequences of the identified pathogenic variants, we performed a detailed structural and conformational analysis of the GUSB protein, with particular emphasis on residues 566 and 388. A comparative evaluation of the wild-type and mutant structures revealed notable alterations in intermolecular interactions, highlighting the structural impact of these substitutions.
At position 566, the wild-type (Leu) protein exhibited two conventional hydrogen bonds, whereas the mutant (Pro) form retained only one, indicating a reduction in stabilizing interactions (Figure A). Two weak hydrogen bonds were preserved in both forms, and neither structure displayed water-mediated hydrogen bonds. Additionally, halogen bonds, ionic interactions, and metal-coordinated contacts were absent in both the wild-type and mutant configurations.
More pronounced differences were observed in non-covalent stabilizing interactions. Aromatic interactions, which play a critical role in maintaining structural stability, were present in the wild-type structure (eight interactions) but were completely absent in the mutant. Similarly, hydrophobic interactions, essential for maintaining protein core integrity, were dramatically reduced from 17 in the wild type to only 1 in the mutant. No carbonyl interactions were detected in either form.
A detailed structural comparison was also performed for residue Tyr388Cys to evaluate the conformational consequences of the mutation. The interaction profile of the wild-type and mutant structures revealed substantial differences, highlighting the destabilizing nature of this substitution. In the wild-type protein, residue 388 formed two conventional hydrogen bonds, whereas only one hydrogen bond was retained in the mutant form, indicating a reduction in stabilizing interactions. Both structures exhibited two weak hydrogen bonds, and no water-mediated hydrogen bonds were detected in either configuration. Additionally, ionic interactions, halogen bonds, and metal-coordination interactions were absent in both the wild-type and mutant structures.
More striking differences were observed in non-covalent stabilizing interactions. The wild-type residue participated in eight aromatic interactions, all of which were completely lost in the mutant structure. Given that aromatic contacts contribute significantly to maintaining tertiary structural stability, their absence suggests a marked weakening of the local structural framework. Similarly, hydrophobic interactions were dramatically reduced from 17 in the wild type to only one in the mutant. As hydrophobic contacts are essential for preserving protein core integrity and conformational stability, this substantial reduction indicates severe structural disruption. No carbonyl interactions were observed in either form.
Overall, the mutation at residues 388 and 566 results in pronounced alterations in hydrogen bonding, aromatic stacking, and hydrophobic interactions, key forces that govern protein stability and structural coherence. These changes strongly suggest that the mutation compromises the equilibrium and functional integrity of the GUSB protein.
4. Conclusions
This study provides a comprehensive characterization of pathogenic nsSNPs in the GUSB gene and elucidates their potential molecular consequences. By systematically identifying deleterious, structurally destabilizing, and disease-associated mutations, we gain critical insight into the mechanistic basis of GUSB-related disorders. The integration of aggregation and solubility analyses further underscores the role of protein solubility in disease progression and highlights how specific amino acid substitutions can promote structural instability and functional impairment. The findings of this study may contribute to the development of targeted therapeutic strategies to mitigate the effects of harmful mutations. Potential approaches include designing small molecules or pharmacological chaperones that stabilize the mutant protein, enhance its solubility, or prevent aberrant aggregation. Moreover, understanding mutation-induced structural alterations may facilitate the rational design of precision therapeutics that restore lost enzymatic function or compensate for structural deficiencies. In conclusion, this integrative analysis of nsSNPs in the GUSB gene advances our understanding of the genetic and structural determinants of disease. By combining sequence-based prediction, structural stability assessment, pathogenicity evaluation, and solubility analysis, this work establishes a robust framework for variant prioritization and lays the foundation for future experimental validation and therapeutic development.
Conflicts of Interest
The authors declare no conflict of interest.
Funding
This work received no funding.
Data Availability Statement
All data generated or analyzed during this study are included in this manuscript.
Declaration on the Use of AI Tools
The authors declare that ChatGPT (OpenAI) was used solely to refine the language, improve grammar, and enhance the clarity of the manuscript.
Figures
Figure 1. Number of SNPs in GusB represented using the dbSNP database.
{kind=link}
Figure 2. A recap of the computational approaches used to anticipate harmful mutations in the GUSB at both structural and functional levels.
{kind=link}
Figure 3. Distribution of deleterious and neutral nsSNPs predicted by sequence-based tools for the entire sequence of GUSB gene.
{kind=link}
Figure 4. Distribution of destabilizing nsSNPs predicted by structure-based tools for the entire sequence of GUSB gene.
{kind=link}
Figure 5. Conserved region of GusB.
{kind=link}
Figure 6. Analysis of Specific mutation (A). Leucine residue 566 mutated to Proline and (B). Tyrosine to Cysteine at position 388.
{kind=link}
References
- 1
Adzhubei, I., Jordan, D.M., Sunyaev, S.R. 2013, Current Protocols in Human Genetics, Chapter 7, Unit7.20, doi: 10.1002/0471142905.hg0720s76 DOI
- 2
Adzhubei, I.A., Schmidt, S., Peshkin, L., et al. 2010, Nature Methods, 7, 248–249, doi: 10.1038/nmeth0410-248 DOI
- 3
Ashkenazy, H., Abadi, S., Martz, E., et al. 2016, Nucleic Acids Research, 44, W344–W350, doi: 10.1093/nar/gkw408 DOI
- 4
Awolade, P., Cele, N., Kerru, N., et al. 2020, European Journal of Medicinal Chemistry, 187, 111921, doi: 10.1016/j.ejmech.2019.111921 DOI
- 5
Balch, W.E., Morimoto, R.I., Dillin, A., Kelly, J.W. 2008, Science, 319, 916–919, doi: 10.1126/science.1141448 DOI
- 6
Bhattacharyya, S., & Tobacman, J.K. 2023, bioRxiv, 2023.04.03.535377, doi: 10.1101/2023.04.03.535377 DOI
- 7
Capriotti, E., Calabrese, R., Casadio, R. 2006, Bioinformatics, 22, 2729–2734, doi: 10.1093/bioinformatics/btl423 DOI
- 8
Capriotti, E., Calabrese, R., Fariselli, P., et al. 2013, BMC Genomics, 14, S6, doi: 10.1186/1471-2164-14-S3-S6 DOI
- 9
Casale, J., & Crane, J.S. 2024, Biochemistry, Glycosaminoglycans
- 10
Chen, Y., Lu, H., Zhang, N., et al. 2020, PLoS Computational Biology, 16, e1008543, doi: 10.1371/journal.pcbi.1008543 DOI
- 11
Choudhury, A., Mohammad, T., Samarth, N., et al. 2021, Scientific Reports, 11, 10202, doi: 10.1038/s41598-021-89450-7 DOI
- 12
Ciryam, P., Tartaglia, G.G., Morimoto, R.I., et al. 2013, Cell Reports, 5, 781–790, doi: 10.1016/j.celrep.2013.09.043 DOI
- 13
Cubizolle, A., Serratrice, N., Skander, N., et al. 2014, Molecular Therapy, 22, 762–773, doi: 10.1038/mt.2013.283 DOI
- 14
D'Avanzo, F., Rigon, L., Zanetti, A., Tomanin, R. 2020, International Journal of Molecular Sciences, 21, 1258, doi: 10.3390/ijms21041258 DOI
- 15
Doherty, G.G., Ler, G.J.M., Wimmer, N., et al. 2023, ChemBioChem, 24, e202200619, doi: 10.1002/cbic.202200619 DOI
- 16
Dubot, P., Sabourdy, F., Plat, G., et al. 2019, International Journal of Molecular Sciences, 20, 5345, doi: 10.3390/ijms20215345 DOI
- 17
Florindo, R.N., Souza, V.P., Mutti, H.S., et al. 2018, New Biotechnology, 40, 218–227, doi: 10.1016/j.nbt.2017.08.012 DOI
- 18
Garantziotis, S., & Savani, R.C. 2022, American Journal of Physiology - Cell Physiology, 323, C202–C214, doi: 10.1152/ajpcell.00088.2022 DOI
- 19
Golda, A., Jurecka, A., Tylki-Szymanska, A. 2012, International Journal of Cardiology, 158, 6–11, doi: 10.1016/j.ijcard.2011.06.097 DOI
- 20
Grubb, J.H., Vogler, C., Sly, W.S. 2010, Rejuvenation Research, 13, 229–236, doi: 10.1089/rej.2009.0920 DOI
- 21
Hua, S.H., Viera, M., Yip, G.W., Bay, B.H. 2022, Cancers, 15, 266, doi: 10.3390/cancers15010266 DOI
- 22
Hubbard, T., Barker, D., Birney, E., et al. 2002, Nucleic Acids Research, 30, 38–41, doi: 10.1093/nar/30.1.38 DOI
- 23
Hytonen, M.K., Arumilli, M., Lappalainen, A.K., et al. 2012, PLoS ONE, 7, e40281, doi: 10.1371/journal.pone.0040281 DOI
- 24
Jefferson, R.A. 1989, Nature, 342, 837–838, doi: 10.1038/342837a0 DOI
- 25
John, R.M., Hunter, D., Swanton, R.H. 1990, Archives of Disease in Childhood, 65, 746–749, doi: 10.1136/adc.65.7.746 DOI
- 26
Khan, S.A., Peracha, H., Ballhausen, D., et al. 2017, Molecular Genetics and Metabolism, 121, 227–240, doi: 10.1016/j.ymgme.2017.05.016 DOI
- 27
Knowles, T.P.J., Vendruscolo, M., Dobson, C.M. 2014, Nature Reviews Molecular Cell Biology, 15, 384–396, doi: 10.1038/nrm3810 DOI
- 28
Kong, X., Zheng, Z., Song, G., et al. 2022, Frontiers in Immunology, 13, doi: 10.3389/fimmu.2022.876048 DOI
- 29
Laimer, J., Hofer, H., Fritz, M., et al. 2015, BMC Bioinformatics, 16, 116, doi: 10.1186/s12859-015-0548-6 DOI
- 30
Landrum, M.J., Lee, J.M., Riley, G.R., et al. 2014, Nucleic Acids Research, 42, D980–D985, doi: 10.1093/nar/gkt1113 DOI
- 31
Leiro, B., Phillips, D., Duiker, M., et al. 2021, Orphanet Journal of Rare Diseases, 16, 500, doi: 10.1186/s13023-021-02113-8 DOI
- 32
Luderschmidt, Ch., Schill, W.-B., Burg, D., et al. 1979, DMW - Deutsche Medizinische Wochenschrift, 104, 1482–1487, doi: 10.1055/s-0028-1129127 DOI
- 33
Nagpal, R., Goyal, R.B., Priyadarshini, K., et al. 2022, Indian Journal of Ophthalmology, 70, 2249, doi: 10.4103/ijo.IJO_425_22 DOI
- 34
Naz, H., Islam, A., Waheed, A., et al. 2013, Rejuvenation Research, 16, 352–363, doi: 10.1089/rej.2013.1407 DOI
- 35
Ng, P.C., & Henikoff, S. 2003, Nucleic Acids Research, 31, 3812–3814, doi: 10.1093/nar/gkg509 DOI
- 36
Page, C. 2013, ISRN Pharmacology, 2013, 910743, doi: 10.1155/2013/910743 DOI
- 37
Paladin, L., Piovesan, D., Tosatto, S. 2017, Nucleic Acids Research, 45, doi: 10.1093/nar/gkx412 DOI
- 38
Pires, D.E.V., Blundell, T.L., Ascher, D.B. 2016, Scientific Reports, 6, 29575, doi: 10.1038/srep29575 DOI
- 39
Rodrigues, C.H., Pires, D.E., Ascher, D.B. 2018, Nucleic Acids Research, 46, W350–W355, doi: 10.1093/nar/gky300 DOI
- 40
Sawamoto, K., Alvarez Gonzalez, J.V., Piechnik, M., et al. 2020, International Journal of Molecular Sciences, 21, 1517, doi: 10.3390/ijms21041517 DOI
- 41
Seker Yilmaz, B., Davison, J., Jones, S.A., Baruteau, J. 2021, Journal of Inherited Metabolic Disease, 44, 129–147, doi: 10.1002/jimd.12316 DOI
- 42
Sherry, S.T., Ward, M.H., Kholodov, M., et al. 2001, Nucleic Acids Research, 29, 308–311, doi: 10.1093/nar/29.1.308 DOI
- 43
Shihab, H.A., Gough, J., Cooper, D.N., et al. 2013, Human Mutation, 34, 57–65, doi: 10.1002/humu.22225 DOI
- 44
Shihab, H.A., Rogers, M.F., Gough, J., et al. 2015, Bioinformatics, 31, 1536–1543, doi: 10.1093/bioinformatics/btv009 DOI
- 45
Shim, S.-B., Kim, N.-J., Kim, D.-H. 2000, Planta Medica, 66, 40–43, doi: 10.1055/s-2000-11109 DOI
- 46
Sim, N.-L., Kumar, P., Hu, J., et al. 2012, Nucleic Acids Research, 40, W452–W457, doi: 10.1093/nar/gks539 DOI
- 47
Staudt, C., Puissant, E., Boonen, M. 2016, International Journal of Molecular Sciences, 18, 47, doi: 10.3390/ijms18010047 DOI
- 48
Stenson, P.D., Mort, M., Ball, E.V., et al. 2009, Genome Medicine, 1, 13, doi: 10.1186/gm13 DOI
- 49
Thal, D.R., Walter, J., Saido, T.C., Fandrich, M. 2015, Acta Neuropathologica, 129, 167–182, doi: 10.1007/s00401-014-1375-y DOI
- 50
Tomatsu, S., Montano, A.M., Oikawa, H., et al. 2011, Current Pharmaceutical Biotechnology, 12, 931–945, doi: 10.2174/138920111795542615 DOI
- 51
Urayama, A., Grubb, J.H., Sly, W.S., Banks, W.A. 2004, Proceedings of the National Academy of Sciences of the United States of America, 101, 12658–12663, doi: 10.1073/pnas.0405042101 DOI
- 52
Wallace, B.D., Roberts, A.B., Pollet, R.M., et al. 2015, Chemistry & Biology, 22, 1238–1249, doi: 10.1016/j.chembiol.2015.08.005 DOI
- 53
Wan, L.M., Zhang, S.K., Li, S.B., et al. 2020, Thrombosis and Haemostasis, 120, 647–657, doi: 10.1055/s-0040-1705117 DOI
- 54
Wang, M., Liu, X., Lyu, Z., et al. 2017, Colloids and Surfaces B: Biointerfaces, 150, 175–182, doi: 10.1016/j.colsurfb.2016.11.022 DOI
- 55
Wei, H., Wang, W., Peng, Z., Yang, J. 2024, Genomics, Proteomics & Bioinformatics, 22, qzae001, doi: 10.1093/gpbjnl/qzae001 DOI
- 56
Yang, G., Ge, S., Singh, R., et al. 2017, Drug Metabolism Reviews, 49, 105–138, doi: 10.1080/03602532.2017.1293682 DOI
- 57
Zhou, Z.H. 2011, Advances in Protein Chemistry and Structural Biology, 82, 1–35, doi: 10.1016/B978-0-12-386507-6.00001-4 DOI
Tables
Table 1. Disease phenotype analysis of high-confidence nsSNPs in the GUSB gene.
| S. No. | Mutations | PhD-SNP | SNP & GO | MutPred2 |
|---|---|---|---|---|
| 1 | I499M | Neutral | Neutral | 0.62 |
| 2 | L121V | Neutral | Neutral | 0.77 |
| 3 | L214P | Disease | Disease | 0.953 |
| 4 | L566P | Disease | Disease | 0.888 |
| 5 | N135D | Disease | Disease | 0.815 |
| 6 | P108L | Disease | Disease | 0.8 |
| 7 | P196H | Neutral | Disease | 0.701 |
| 8 | R36L | Disease | Disease | 0.943 |
| 9 | R398H | Disease | Neutral | 0.561 |
| 10 | R56C | Disease | Neutral | 0.361 |
| 11 | T180P | Disease | Disease | 0.843 |
| 12 | T226P | Disease | Disease | 0.497 |
| 13 | V601G | Disease | Neutral | 0.545 |
| 14 | Y388C | Disease | Disease | 0.785 |
| 15 | Y399C | Disease | Disease | 0.567 |
Table 2. Prediction of aggregation propensity of mutant GUSB using SODA server.
| Mutations | Sequence | Helix | Strand | Aggregation | Disorder | SODA | Result |
|---|---|---|---|---|---|---|---|
| Wild type | 0.281 | 0.296 | -5.254 | 0.024 | |||
| L214P | 1 | -1.351 | 0.165 | 18.495 | 0.024 | 16.507 | More soluble |
| L566P | 2 | -2.1 | 0.568 | 5.083 | 0.034 | 2.178 | More soluble |
| N135D | 3 | 2.394 | -2.928 | 116 | 0.003 | 115.555 | More soluble |
| P108L | 4 | 0.707 | -0.875 | 12.252 | 0.004 | 11.977 | More soluble |
| R36L | 5 | -2.585 | 2.598 | -0.668 | -0.445 | -2.167 | Less soluble |
| T180P | 6 | -0.248 | -0.335 | 17.006 | 0.149 | 16.553 | More soluble |
| Y388C | 7 | -1.912 | 1.187 | -0.577 | -0.033 | -1.992 | Less soluble |
| Y399C | 8 | 0.028 | 0.82 | -22.711 | -0.069 | -21.467 | Less soluble |