


About Article
Genome-wide annotation and structural modeling of hypothetical proteins in Listeria monocytogenes
Abstract
The rapid expansion of genome sequencing projects has resulted in the identification of numerous hypothetical proteins whose functions remain uncharacterized. In Listeria monocytogenes serotype 4b, a major food-borne pathogen associated with high mortality rates, several predicted proteins lack functional annotation despite their potential role in pathogenicity and survival. In the present study, a comprehensive in silico approach was employed to functionally annotate 92 hypothetical proteins identified from the genome of L. monocytogenes. Protein sequences were retrieved and analyzed using sequence similarity searches, conserved domain identification, motif analysis, and multiple sequence alignment. Functional classification was performed based on BLAST, Pfam, and InterPro analyses. Structural prediction was performed via homology modeling via the SWISS-MODEL server, followed by structural validation and comparative analysis using PyMOL and DALI-Lite. Functional inference from structural models was supported by conserved-residue mapping and ProFunc analysis. Sequence-based analysis enabled classification of most hypothetical proteins into functional groups, including hydrolases, transferases, transporters, kinases, stress-response proteins, membrane proteins, DNA-binding proteins, and ATP-binding proteins. Structure-based modeling of selected proteins further confirmed the predicted catalytic residues, metal-binding sites, ligand-interaction sites, and conserved functional motifs. Several proteins were predicted to be involved in enzymatic activity, nucleotide metabolism, membrane transport, transcriptional regulation, and stress adaptation. This integrative computational analysis provides functional insights into previously uncharacterized proteins of L. monocytogenes. The findings enhance genome annotation quality and identify potential targets for further experimental validation, contributing to a better understanding of bacterial physiology and pathogenesis.
Keywords
1. Introduction
Due to the speed and cost-effectiveness of genome sequencing, many small bacterial, archaeal, and eukaryotic genomes have been sequenced, and larger eukaryotic genomes are expected to be fully sequenced soon. In such a case, annotation becomes problematic when genes are predicted faster than annotation can keep pace. Despite ongoing improvements in genome annotation, a substantial proportion of open reading frames remain classified as ``conserved hypothetical proteins.'' Hypothetical proteins are predicted from nucleotide sequences but lacking direct experimental validation at the protein level (?). In many instances, these predicted proteins show limited sequence similarity to previously characterized and annotated proteins. Conserved hypothetical proteins constitute a significant fraction of genes in sequenced genomes; they encode proteins identified across multiple phylogenetic lineages but remain uncharacterized with respect to biological function or biochemical properties, sometimes accounting for a large portion of the predicted proteome (?).
Determining the functions of hypothetical proteins is essential for improving genomic and proteomic annotations. The identification of new hypothetical proteins can reveal previously unknown structural folds and biological activities (?). As more structures are characterized, novel domains, motifs, and conformational arrangements are likely to emerge, contributing to a clearer understanding of structure–function relationships in proteins (?). Functional characterization may also uncover new molecular pathways and regulatory cascades. Furthermore, hypothetical proteins may serve as biomarkers or therapeutic targets. Their identification can facilitate the discovery of previously unrecognized or computationally predicted genes (?). Ultimately, elucidating the roles of these proteins will deepen our understanding of biological systems and may contribute to the development of improved therapeutic interventions (?; Rao, 1989).
The biological roles of many predicted proteins remain unknown because their existence has been inferred solely from genomic sequences (?). The rapid completion of genome sequencing projects has generated extensive biological datasets, enabling the association of genes with their corresponding protein products, which are fundamental to cellular function. Advances in genome and cDNA sequencing technologies allow the prediction of numerous proteins for which experimental evidence is lacking (Gury et al., 2004). Functional insights into hypothetical proteins are often derived from sequence comparisons with proteins of known function in model organisms. Since similarities in sequence or conserved domains frequently indicate shared functional properties, such analyses can suggest potential biological roles (?). Moreover, if a hypothetical protein exhibits significant structural homology with characterized proteins, it is reasonable to infer comparable molecular features (?).
Traditional biochemical and molecular biology approaches can accurately assign functions to genes; however, these experimental methods are labor-intensive and costly. Consequently, even in fully sequenced genomes, only about 50–60% of genes have been functionally annotated (?). Automated genome analysis and computational annotation strategies provide alternative means to interpret genomic information. Therefore, determining protein function remains a major challenge in the post-genomic era. This has increased reliance on bioinformatics tools to predict the roles of uncharacterized protein sequences, and several contemporary computational approaches have been developed to address this issue (?).
Listeria monocytogenes is the causative agent of listeriosis. It is a facultative anaerobic bacterium capable of surviving under both aerobic and anaerobic conditions (?). The organism can invade and proliferate within host cells and is recognized as one of the most severe foodborne pathogens, with mortality rates of 20–30% in clinical cases (?). In the United States, listeriosis is responsible for approximately 2,500 reported cases and 500 deaths annually, making it one of the leading causes of death among foodborne bacterial infections (?). Although relatively rare, the disease is associated with a high case-fatality rate. Its emergence as a public health concern has been attributed to changing dietary habits, advances in food preservation technologies that extend shelf life, and the bacterium’s ability to survive and grow at refrigeration temperatures.
Disease caused by Listeria monocytogenes is called Listeriosis (a bacterial infection). In most cases, these infections occur in immunocompromised individuals (?), such as pregnant women (they are about 20 times more likely to get infected than healthy adults), Newborns, persons with diabetes, cancer, and kidney disease, persons with AIDS, and persons who take glucocorticosteroid medications. Listeriosis is fatal in at least 1 in 5 infected individuals. Listeria monocytogenes causes flu-like symptoms and meningitis if it spreads to the central nervous system. The first reported case of Listeriosis was in 1981; since then, numerous listeriosis outbreaks have been detected and investigated. Subsequent studies have confirmed that the Listeria monocytogenes serovar primarily responsible for Listeria infection is serotype 4b. So, of the 13 serovars of Listeria monocytogens, Listeria monocytogenes 4b is the most prominent one (?). Listeria monocytogenes has been reported to cause miscarriages, stillbirths, or even premature pregnancies among pregnant women with underlying diseases. The chances of a fetus being affected by an affected mother are high.
The Listeria monocytogenes genome is approximately 3 Mb. Its genome consists of one chromosome. Total number of nucleotides is 2912690, total number of protein genes found to be 2766, and total number of RNA genes is 85. Many stress proteins are expressed in Listeria monocytogenes under stressful conditions, helping to bacterium survive harsh environments for extended periods. There are several important facts about Listeria monocytogenes genome available. Genome analysis suggests that the Listeria monocytogenes 4 b genome contains several hypothetical proteins. There are 92 hypothetical proteins in Listeria monocytogenes 4 b. There is little information available on these hypothetical proteins from Listeria monocytogenes genome. Here, our aim is to annotate the hypothetical proteins from Listeria monocytogenes by using recent bioinformatics tools. After annotating a genome, we grouped genes into two types: the first comprises known genes with functional characterization; the second comprises conserved hypothetical genes conserved across many organisms. Generally, a newly sequenced genome contains approximately 30% of genes that are poorly or incorrectly annotated (?). Hence, these poorly characterized hypothetical genes might play an important role in our understanding of the pathogen's pathogenesis and virulence. Given that so little is known about these genes, we have attempted to assign plausible functions to hypothetical proteins. The functional evidence suggests that these hypothetical proteins may have specific functions, but further detailed analysis to needed to confirm their functions.
2. Materials and Methods
2.1. Data mining
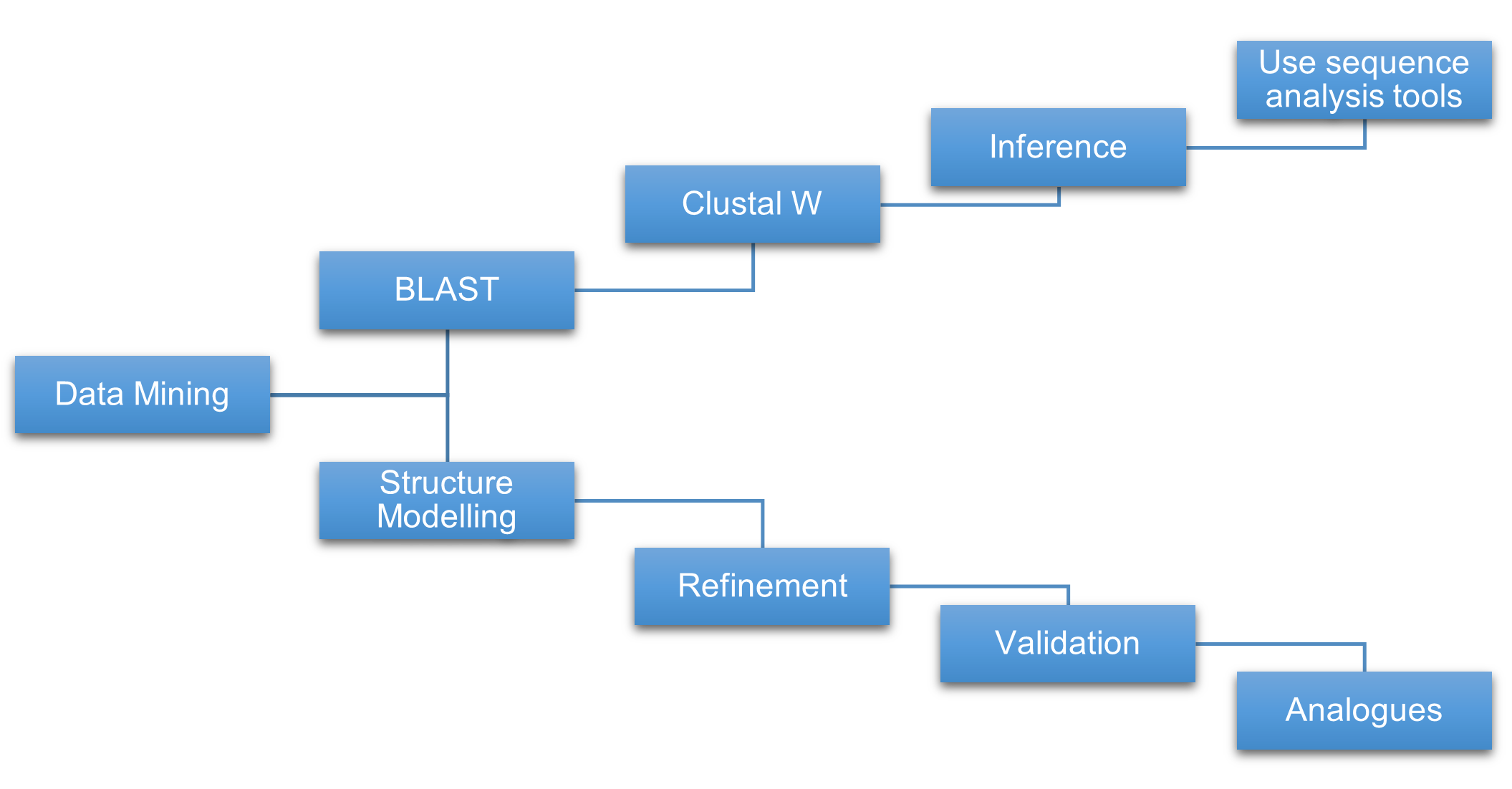
We have analyzed the Listeria monocytogenes's genome and the hypothetical proteins that it contains by retrieving the information at the P1R website. We have identified and downloaded 92 hypothetical protein sequences of Listeria monocytogenes. The sequences of all proteins analyzed with their primary accession number, length, molecular mass, and proposed functions (Table –). Protein sequences are saved in FASTA format for further processing. A systematic pipeline used in this study is illustrated in Figure.
2.2. Sequence similarity search
We have derived the function of hypothetical proteins from sequence similarity to a well-characterized homolog in GenBank. Standard protein-protein BLAST (BLASTP) is used to identify a query amino acid sequence and to find similar sequences in protein databases. The results of a BLAST query against a public database provide insight into the functional properties of related sequences. Hence, BLASTP revealed several related searches for our hypothetical proteins. With the help of multiple sequence alignments (ClustalW) of the related selected sequences, we have been able to analyze the conserved residues present in our hypothetical protein by comparing with the proteins of known function, and we have related the functions of the conserved positions of some residues. A multiple sequence alignment was performed using ClustalW to study conserved sequence patterns and ancestral relationships among the organisms. The next step was to predict the functions using structural analysis.
2.3. Structure Prediction
The three-dimensional structures of hypothetical proteins were examined to infer their potential biological functions. Structural prediction was carried out using homology modeling, which generally involves multiple sequential steps. First, a suitable template protein with an experimentally determined structure, typically belonging to the same or a closely related family, is identified. Second, appropriate sequence alignment tools are employed to obtain the best possible alignment between the target and template proteins. Third, the target sequence is divided into smaller segments to facilitate accurate modeling. Fourth, corresponding fragments are searched in structural databases based on sequence similarity and the spatial conformation of the selected template. Fifth, the spatial coordinates of these matched segments are assembled and adjusted to construct the full-length target structure, ensuring that all atomic positions are properly incorporated. These procedures are repeated multiple times to generate several models, from which an averaged structure is obtained, followed by comprehensive energy minimization to refine the final predicted model. The 3-D structures were retrieved using the SWISS-MODEL server, an automated comparative modeling system for predicting protein 3-D structures. The entire homology modeling workflow can be performed in the `project mode' of PyMOL, an integrated sequence-structure workbench. Using the SWISS-MODEL server, we obtained suitable 3D structures for thirteen hypothetical proteins selected for detailed structural analysis, and the resulting structural models were downloaded in PDB format for further analysis.
2.4. Function prediction
Clustering of gene expression profiles is widely used for functional prediction, based on the principle that genes involved in related biological processes tend to exhibit coordinated expression patterns. Functional inference can also be derived from protein–protein interaction networks, where the role of an uncharacterized protein is estimated from the known functions of its interacting partners. Schwikowski and co-workers introduced a neighbor-counting strategy in which the probable function of an unknown protein is determined according to the frequency of functional annotations among its interaction partners. This strategy was later improved by Hishigaki et al.\ (2001) through the incorporation of a chi-square (chi2) statistical framework to enhance prediction reliability. In both methods, all functional contributions from neighboring proteins are treated with equal significance. Several publicly accessible signature and motif databases support functional annotation and are frequently integrated with sequence clustering and domain-based resources. These include PROSITE, PRINTS, Pfam, ProDom, Blocks, SMART, and InterPro; in the present study, Pfam and InterPro were specifically utilized. Structural classification of proteins into homologous families, superfamilies, and folds can be performed using databases such as SCOP, CATH, FSSP, CAMPASS, and HOMSTRAD. Additionally, the ProFunc server facilitates structure-based functional prediction by comparing modeled protein structures against multiple databases and reporting significant matches for functional interpretation.
2.5. Visualization
We carried out structural analysis using PyMOL for superposition of molecules, secondary-structure-based alignment, and calculation of RMSD values. PyMOL is also used to depict structures, as well as to analyze potential residue positions and functions. We visualized the template residues and the model after superimposing the model onto the template. Each residue was visualized individually, and residue conservation was assessed.
2.6. Structure analysis
Structure-based comparison approaches are highly effective for detecting remote evolutionary relationships that may not be apparent from sequence similarity alone. Structural information also enables the recognition of functional sites that have arisen independently during evolution. Because protein function is inherently dependent on three-dimensional conformation, structural analysis provides direct insight into the molecular mechanisms underlying biological activity. Local structural alignment focuses on identifying conserved three-dimensional arrangements of specific residues, even among proteins that differ substantially in overall fold. Several computational tools are available for such analyses, including DaliLite, which utilizes Cα–Cα distance matrix comparisons; TOPS; SSM; VAST; GRATH, which represents secondary structure elements as graph nodes connected by spatial relationships; and SSAP, which evaluates structural similarity based on Cα comparisons and additional scoring features. Other methods such as LSQMAN, CE (Combinatorial Extension), MATRAS, and LOCK2 also employ atom-based or secondary structure vector representations for alignment. In the present study, DALI-Light was selected for structural comparison and analysis.
3. Results and Discussion
3.1. Sequence-based Function Prediction
Information derived from BLAST, ClustalW, and other online servers for sequence analysis revealed close similarities of hypothetical proteins to proteins of known function. Based on the proposed functions, we classified all 92 hypothetical proteins into functional classes. All the Hypothetical proteins have been classified into the following categories.
Hydrolase
The enomic proteins of Listeria monocytogenes most abundantly showed similarity with proteins having hydrolase function, and hence, by comparing the conserved residues, we can assume the proteins to have the same function as well. The sequence of C1L0U3, C1L0R8, C1KZQ3, C1KZE1, C1KYZ2, C1KYY1, C1KY45, C1KY43, and C1KXZ5 has been predicted to have hydrolase function following a precise analysis that compared its sequence similarity with that of known, characterized proteins. Hydrolases are enzymes that facilitate hydrolytic reactions, in which a water molecule is utilized to break chemical bonds within a substrate. During this process, the hydrogen (H+) and hydroxyl (OH-) components of water are incorporated into the reacting molecule, resulting in its cleavage into two or more smaller products. The term ``hydrolase'' serves as the systematic designation for enzymes classified under Enzyme Commission (EC) class 3, which encompasses all enzymes that catalyze hydrolysis reactions.
For the C1L0U3 gene, 65 matching sequences were found by FASTA search. 8 neighboring genes found as Q8Y970, D2PAB8, D2NZC4, C1L0U3, E1UE28, B8DIC3, Q92DZ3, D3UKJ4 having the percentage identity of 100%, 100%, 100%, 100%, 99.6%, 99.6%, 99.3%, 91.5% respectively. 10 sequence motifs were identified when the sequence was searched against the InterPro database. There are 57 matching sequences found by FASTA search for C1KZQ3 gene and 7 neighboring genes were found as E1UBT1, B8DAU2, Q8Y3Y0, D2P7B6, D2NVU7, Q927E2, D3USV7 with identities as 99.3%, 99.3%, 97.8%, 96.8%, 96.8%, 95.3% and 83.5% respectively. 9 sequence motifs were found by using InterPro database. For the C1KY43 gene, we identified 47 matching sequences via a FASTA search and 9 sequence motifs via InterProScan. E1UAS5, B8DDF3, Q8Y4N4, D2P959, D2NY72, Q928N2, and D3UR85 are the 7 neighbouring genes, with percentage identities of 98%, 97.6%, 97.2%, 97.2%, 97.2%, 95.6%, and 94%, respectively.
Transferase
Transferases are enzymes that catalyze the movement of specific chemical groups, such as methyl, glycosyl, acyl, or phosphate groups, from one molecule to another. In these reactions, one compound acts as the donor of the functional group, while another serves as the acceptor. The nomenclature and classification of transferases follow the format ``donor:acceptor group transferase,'' reflecting the nature of the transferred group and the participating substrates. Proteins belonging to this group are C1L2K8, C1L1Z7, C1L0P3, C1L0L0, C1L024, C1KYI4, C1KXY4, C1KVW1, and C1KVA0. The protein sequence has been found to be similar to that of characterized proteins in this category. This group is the second most abundant in Listeria monocytogenes, after hydrolases. For the gene C1L0L0, 65 matching sequences were identified by a FASTA search, and 8 motifs were matched by InterProScan, which searches the InterPro database. The 7 neighbouring genes are as follows D2PA32, D2NYL3, Q8Y9E8, E1UDU6, B8DA44, Q92E70 and D3USH9 with percentage identity as 99.5%, 99.5%, 99.5%, 96.7%, 96.7%, 92.8% and 89.4% respectively. There are 29 matching sequences found by FASTA search for the C1KXY4 gene and 6 motifs matched while running it against InterPro database. There are 6 neighboring genes found which are E1UCN5, B8DGQ6, Q8YAG1, D2PAN3, D2NZA9 and Q92F98 with percentage identity as 97.1%, 97.1%, 95.5%, 95.1%, 95.1% and 95.9% respectively.
Transporter
Transporter proteins are proteins that move substances within an organism. Transport proteins are vital to the growth and life of all living things. There are several different kinds of transport proteins. The proteins found in Listeria monocytogenes included in this category are C1L300, C1L1R1, C1L164, C1L0K2, C1KZ81, C1KZ17, C1KYZ6, C1KY61, and C1KY46. For the C1L1R1 gene, 4 matching sequences were found by FASTA search, and 6 motifs were found by InterPro scan. There were 7 neighboring genes found which are E1UF08, B8DEE5, Q8Y8B8, D2P3J7, D2P0R1, Q92D31, and D3ULQ9 with percentage identity as follows 97.9%, 97.9%, 96.8%, 96.6%, 96.6%, 93.4%, and 87.4%, respectively.
RNA Binding
The following residues belong to this group: C1L2S2, C1L1U8, and C1L1G8. RNA-binding proteins are proteins that bind to RNA recognition motifs (RRMs) of double or single-stranded RNA in cells and participate in forming ribonucleoprotein complexes. They are cytoplasmic and nuclear proteins. For the C1L2S2 gene, 15 matching sequences were identified by FASTA search, and 11 sequence motifs were identified in InterPro. The 7 neighbouring genes are Q8Y7C0, D2P4S6, D2P1Z0, E1UG75, B8DFW2, Q92BY9 and D3UMV0 with sequence identity as 97.4%, 97.4%, 97.4%, 95.2%, 95.2%, 94.9% and 94.9% respectively. Whereas for the C1L1U8 gene, 51 matching sequences were found, and 11 sequence motifs matched in InterPro. There are 7 neighbouring genes which are as follows Q92CZ5, Q7AP12, E1UF45, D2P3N4, D2P0U8, B8DEA8 and D3ULU6 with similarities as 100%, 100%, 100%, 100%, 100%, 100%, and 99.5% respectively.
Hydrolase acting on carbon-nitrogen but not on peptide
Following mentioned are the proteins C1L161, C1KYM2, and C1KXZ0, which belong to this category. This family contains hydrolases that break carbon-nitrogen bonds. This sub family of hydrolase is numbered as EC 3.5. There were 35 matching sequences found by FASTA search and 6 sequence motifs matched in the InterPro scan. There were 6 neighbouring genes found which are E1UD07, D2P8J8, D2NWP2, B8DEW3, Q8YA77, and Q92EZ8 with percentage identity as 99.2%, 99.2%, 99.2%, 99.2%, 98.1% and 95.3% respectively.
Kinase
Kinases are enzymes that catalyze the transfer of phosphate groups from high-energy donor molecules, typically adenosine triphosphate (ATP), to specific target substrates in a reaction known as phosphorylation. Through this process, kinases regulate numerous cellular activities, including signal transduction, metabolism, and cell cycle progression. They belong to the broader class of phosphotransferases. In the present study, proteins C1L143, C1KZU1, and C1KYX3 were identified as members of this enzyme category.
Response to stress
Cells exposed to stressful conditions often accumulate abnormal, misfolded, or damaged proteins as a consequence of environmental or physiological challenges. To mitigate these effects, cells increase the production of stress-related proteins that assist in stabilizing, refolding, or degrading altered proteins. Rather than solely enabling survival under extreme or lethal stress, these stress proteins also play a crucial role in cellular recovery following stress exposure, thereby helping restore normal cellular homeostasis. C1LO28, C1KZM7, and C1KW03 are the proteins that belong to this category. A FASTA search of the C1KZM7 gene identified 21 matching sequences, and an InterPro scan identified 4 sequence motifs. Q927G8, E1UBQ4, D2P7S2, D2NVX3, B8DAW9, Q8Y405 and D3USS9 are the 7 neighbouring genes found with 99.3%, 99.3%, 99.3%, 99.3%, 99.3%, 98.6% and 98.6% identity respectively.
Integral to membrane
An integral membrane protein is a protein molecule, or protein complex, that is permanently embedded within the plasma membrane through hydrophobic regions that interact strongly with the lipid bilayer. These proteins are tightly associated with membrane phospholipids and cannot be easily removed without disrupting the membrane structure. Integral membrane proteins are broadly divided into two principal categories: (1) transmembrane proteins and (2) integral monotropic proteins. Transmembrane proteins represent the more prevalent group and extend completely across the lipid bilayer. Such proteins perform diverse biological functions, including acting as transporters, ion channels, receptors, enzymes, structural anchors, components involved in energy storage and signal transduction, and mediators of cell–cell adhesion. Representative examples include integrins, cadherins, the insulin receptor, neural cell adhesion molecules (NCAM), selectins, glycophorin, and rhodopsin. C1L1R3, C1L1R2, and C1KY64 are the proteins that belong to this group. For the gene C1KY64, we found 39 matching sequences by FASTA search and 5 motifs matched in InterPro scan. 6 neighbouring genes found were E1UAU6, B8DDD2, Q8Y4L1, D2P980, D2NY93, and Q928L2 with percentage identity as 98.3%, 98.3%, 96.6%, 96.6%, 96.6%, and 97.4%, respectively.
DNA Binding
DNA-binding proteins are proteins that contain a DNA-binding domain, which in turn is responsible for binding to single or double-stranded DNA molecules. C1L0T5 and C1KWG4 are the proteins that have this domain and are therefore classified in this category.
Phosphoesterase
Phosphoesterase is a family of hydrolase with acts on ester bonds. Phosphoesterases are involved in cleaving ester bonds. Proteins belonging to this category are C1L2V7 and C1L2E5. For gene C1L2E5 there are 24 matching sequences found by FASTA search and 6 sequence motifs matched in InterPro. There are 7 neighbouring genes which are Q8Y7N4, E1UFM2, B8DHZ2, D2P485, D2P1E9, Q92CG9 and D3UME0 with percentage identity as 100%, 100%, 100%, 99.4%, 99.4%, 97.6%, and 95.3% respectively.
ATP Binding
An ATP-binding protein is a protein that specifically interacts with adenosine 5'-triphosphate (ATP), a ribonucleotide composed of the purine base adenine attached to the sugar D-ribofuranose and linked to three phosphate groups. ATP serves as a primary carrier of chemical energy and phosphate groups within the cell. Proteins that bind ATP typically utilize this interaction to drive biochemical reactions, regulate cellular processes, or facilitate molecular transport, thereby playing essential roles in energy-dependent cellular functions. C1KYL6 and C1KV71 are the proteins in this category. For the C1KYL6 gene, we identified 69 matching sequences via a FASTA search. D2P8J2, D2NWN6, Q8YA83, Q92F06, D3URG2, E1UCZ8, and B8DEX2 are the 7 neighboring genes found with percentage identities as 98.9%, 98.9%, 98.5%, 90.3%, 89.2%, 89.6%, and 89.6%, respectively. Additionally, 10 sequence motifs were matched in the InterPro scan. For the C1KV71 gene, 102 matching sequences were identified by a FASTA search, and 11 sequence motifs were identified by an InterPro scan. 7 neighboring genes found were E1U8I5, B8DAN0, Q8YAT3, D2PBF4, D2P030, Q92FS4 and D2P8J2 with percentage identity as 96.6%, 96.6%, 96.2%, 96.2%, 96.2%, 95.8% and 50.2% respectively.
Other Proteins
Proteins in this category are proteins that are one of their kind, i.e., no other protein in Listeria monocytogenes shares the same function as they do. Proteins like C1L1B3 whose function is mRNA cleavage, C1L165 whose function is to bind to polyisoprenoid, C1L158 which is responsible for phenazine biosynthesis, C1L0M8 which is involved in negative regulation of phenolic acid metabolism, C1K0G9 whose function is to catabolize myo-inositol, C1L075 which is polyphosphogluconolactonase, C1KZV6 which is responsible for nucleotide binding, C1KYZ4 which is monoxygenase, C1KYX9 which is involved in proteolysis, C1KYT3 is a GTP binding protein, C1KY51 is iron binding protein, C1KY50 has iron-sulphur cluster assembly, C1KY30 has chloride chemical activity, C1KXN7 is an oxidoreductase, C1KWC7 has a role in stationary phase survival, C1KVW8 has catalytic activity. The proteins are among those that don’t share any function with any other protein. This is why they are being classified in this category. For the C1L165 gene, 11 matching sequences were identified by a FASTA search, and 4 sequence motifs were identified by an InterPro scan. There were 7 neighbouring genes found which are as follows Q8Y8U6, E1UEF3, D2PB35, D2NZR0, B8DGH0, Q92DM4 and D3UL61 with percentage identities of 99.4%, 99.4%, 99.4%, 99.4%, 99.4%, 98.3%, and 95.9%. 25 matching sequences were found by a FASTA search for the C1L0M8 gene. Additionally, 4 sequence motifs were identified by InterProScan. 7 neighbouring genes found are Q92E53, Q7AP25, D2PA50, D2NYN1, E1UDW6, B8DA24 and 3UKC6 with percentage identity as 100%, 100%, 100%, 99%, 99%, 99%, and 99% respectively.
Unknown
Proteins included in this category are the proteins whose function could not be predicted due to a lack of any known characterized protein hits. Due to the unavailability of good hits, we classified them in this category, as no function can be assigned to them until we obtain hits of characterized proteins whose function is known to us, instead of hypothetical, uncharacterized, or putative proteins, which we obtained for proteins of this category.
3.2. Structure-based Function Prediction
We have sought to develop a template for building models of hypothetical proteins. Fortunately, we identified templates for 13 proteins and subsequently predicted their structures. To predict the function of hypothetical proteins, we have extensively analyzed the atomic coordinates of models and proposed corresponding functions based on structural similarity, fold, domain, motif, and analysis of structurally conserved residues.
C1L1U8
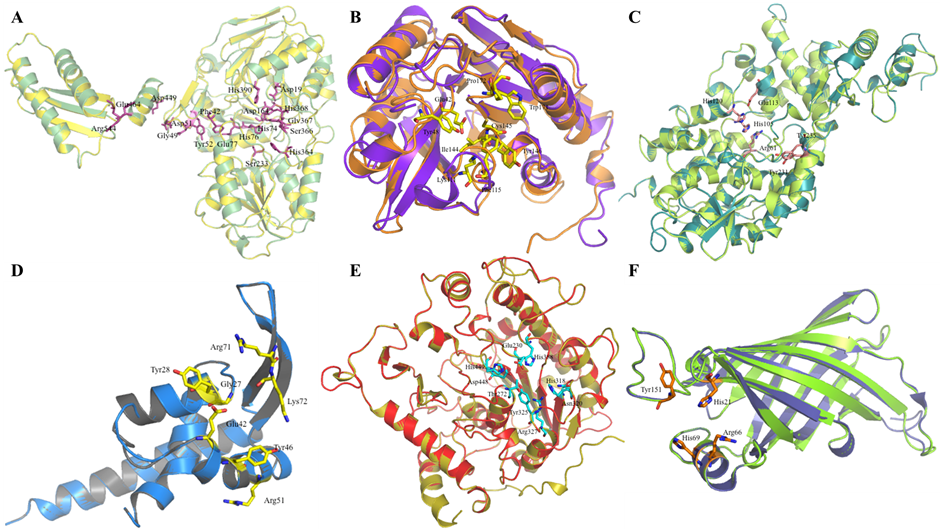
The protein C1L1U8 is 555 residues long, and the model was successfully built for residues 6–555. The overall structure of this protein is shown in Figure A. Our model shares 81.09% sequence identity with the template structure and has an RMSD of 0.1 Å. Furthermore, there were 15 α helices and 23 β sheets in our model. The active site of the template structure 3ZQ4 contains two Zn2+ ions positioned approximately 3 Å\ apart within an octahedral coordination geometry, situated near the cleft separating the β-lactamase and β-CASP domains. The first zinc ion is coordinated by the side chains of Asp78, His79, Asp164, and His390, whereas the second zinc ion interacts with His74, His76, His142, and Asp164 (?). As anticipated, residues involved in metal coordination are highly conserved. Structural superimposition demonstrated that these key residues are preserved in our modeled protein. Additionally, Asp195 and His368 have been implicated in acid–base catalysis. In the template structure, dimerization is stabilized by a Ca2+ ion located at the interface, coordinated by Gly49 and Asp51 from one monomer and Asp443 and Glu464 from the opposing chain. A salt bridge between Asp449 and Arg544 further reinforces the dimer interface. Hydrogen-bonding interactions involve His364, Gly367, Asp78, and Asp164 (?). The residue Ser366 is essential for catalytic activity, and its mutation abolishes enzymatic function. Two conserved residues positioned near the catalytic center, His368 and Asp195, adopt conformations consistent with a classical catalytic dyad. ProFunc analysis identified 20 significant ligand-binding templates among 94,569 entries and one notable DNA-binding template from a dataset of 4,194. The phosphate group of nucleotide 3 forms hydrogen bonds with the side chain of Ser233 and the backbone nitrogen of Glu77. The nucleotide base is oriented to allow potential π–π stacking interactions with Phe42, although optimal stacking requires an alternative rotamer conformation of this residue. Notably, substitution with a pyrimidine base at this position enhances structural compatibility and enables additional hydrogen bonding with the main-chain carbonyl of Asp51 and the hydroxyl group of Tyr52; the structural model was adjusted accordingly to optimize these interactions. Based on the pronounced structural similarity to the template and conservation of catalytic residues, C1L1U8 is proposed to function as a nuclease, possibly exhibiting 5'–3' exonuclease activity. Nevertheless, experimental studies are necessary to validate its exact enzymatic function.
C1KYM2
The protein C1KYM2 comprises 259 residues, and the model was successfully built for residues 2–259. The overall structure of this protein is shown in Figure. Our model shares 37.31% sequence identity with the template and has an RMSD of 1.359 Å. Furthermore, our model contained 6 α-helices and 11 β-sheets. The catalytic triad residues Glu43, Lys109, and Cys143 have been shown to be conserved in our model. Lys 109 and Tyr 144 are responsible for the formation of hydrogen bonds (Chin et al., 2007). In the protein–ligand complex, the two methyl groups of cacodylate establish favorable C–Hcdotsπ interactions with nearby aromatic residues. One methyl group interacts with Phe49, while the other engages Phe113 and Trp175. Such C–Hcdotsπ contacts are recognized as stabilizing forces that can significantly enhance the structural integrity of proteins and protein–ligand assemblies. The binding cavity is largely composed of hydrophobic amino acids, including Phe49, Phe113, Val142, Trp175, and Pro176, which together with residues of the catalytic triad contribute to shaping the active-site pocket (Chin et al., 2007). ProFunc analysis identified 3 statistically significant ligand-binding templates from a total of 94,569 entries examined. Additionally, 2 significant matches were detected among 4,194 DNA-binding templates, suggesting possible nucleic acid interaction capability.
C1KYY1
The protein C1KYY1 comprises 440 residues, and its model was successfully built for residues 1–439. The overall structure of this protein is shown in Figure. Our model shares 48.23% sequence identity with the template structure and has an RMSD of 0.7 Å. Furthermore, there were 21 α helices and 9 β sheets in our model. The residues His-129–Glu-122 form a putative catalytic dyad in the template structure and are among the most important for its activity (Vorontsov et al., 2011). His 66, His 101, Asp 111, and Asp 183 are important for the formation of a bowl-shaped structure where active binding of the ligand takes place. Residues Lys-14, Asn-36, Gln-41, Arg-44, Phe-64, Arg-326, and Lys-330 are in direct contact with the ligand and make this site highly specific to dGTP (Vorontsov et al., 2011). All residues except Asn-36 are highly conserved and play important roles in ligand binding. The His-129, Glu-122 couple has been shown to function as a catalytic dyad for the generation of the nucleophilic OH- hydroxyanion, and a nearby His-114 residue may also assist in the proper positioning of the attacking group. Tyr 239 and Arg 63 form a hydrogen bond (Vorontsov et al., 2011). Leu49–Tyr368 are involved in van der Waals interactions. Because most of the important residues are conserved in our model, we propose that C1KYY1 functions as a deoxynucleotide triphosphate triphosphohydrolase (dNTPase). Furthermore, during ProFunc analysis, 20 significant ligand binding templates were found out of 94569 ligand binding templates.
C1L0M8
The protein C1L0M8 is 110 residues long, and the model was successfully built for residues 5–105. The overall structure of this protein is shown in Figure. Our model shares 40.78% sequence identity with the template and has an RMSD of 0.5 Å. Furthermore, our model contains 4 α-helices and 2 β-sheets. Tyr46 and Arg51 are the DNA-binding residues in the template and help in attaching to DNA. The residues Gly25, Tyr26, Glu42, Arg71, and Lys72 contribute to the strengthening of the binding between DNA and the protein (Fibriansah et al., 2012). Based on the conserved nature of important residues, it can be proposed that the function of C1L0M8 is a transcriptional regulator of phenolic acid.
C1L0R8
The protein C1L0R8 comprises 606 residues, and the model was successfully built for residues 201–605. The overall structure of this protein is shown in Figure. Our model shares 30.07% sequence identity with the template and has an RMSD of 0.5 Å. Furthermore, there are 11 α helices and 9 β sheets in our model. It has been shown that LTA (Lipoteichoic Acid) is required for divalent cation homeostasis and that its absence has severe effects on cell morphogenesis and division. Disruption of LTA synthesis severely affects cell morphology and viability. The residue Thr 297 has been shown to be essential for LTA synthesis and is conserved in our model. Sequence examination of residues directly interacting with the phosphothreonine at position 297, namely His412, Glu253, and Trp350, indicates that these amino acids are highly conserved across LtaS homologs. Their strong conservation among orthologous proteins suggests a critical functional role, likely contributing significantly to lipoteichoic acid (LTA) biosynthesis (?). Glu253, Asp471, and His472, which surround the residue Thr297, have been found to coordinate with the magnesium ion in the active site. His343, Asn345, Arg352, Glu253, and Thr408 are involved in hydrogen-bond formation with water (?).
C1L165
The protein C1L165 is 176 residues long, and the model was successfully built for residues 5–176. The overall structure of this protein is shown in Figure. Our model shares 41.28% sequence identity with the template and has an RMSD of 0.2 Å. Furthermore, our model contains 3 α-helices and 9 β-sheets. His18, Arg62, His65, and Try146 residues are responsible for binding to poly-isoprenoid (Handa et al., 2004). In the ProFunc analysis, 1 significant DNA-binding template was identified among 4194 DNA-binding templates.
C1L2E5
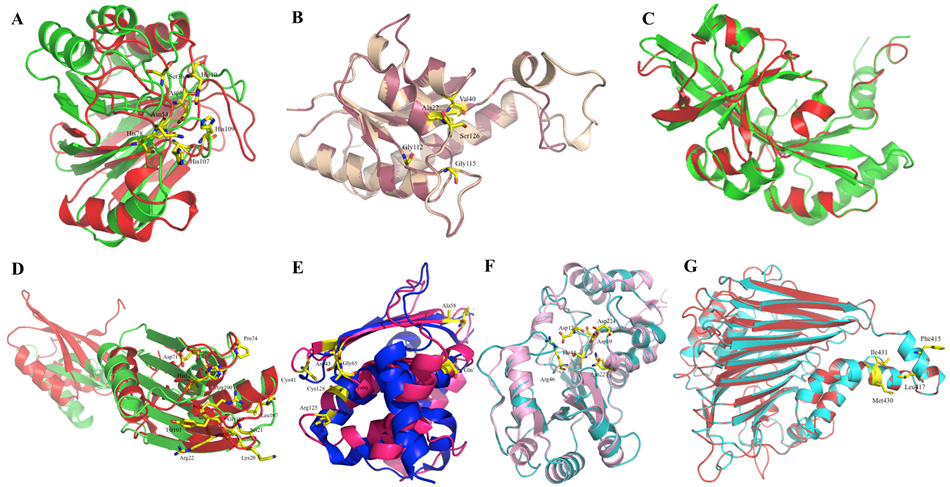
The protein C1L2E5 comprises 169 residues, and the model was successfully built for residues 1–158. The overall structure of this protein is shown in Figure. Our model shares 22.50% sequence identity with the template and has an RMSD of 3.919 Å. Furthermore, our model contains 5 α-helices and 10 β-sheets. Residues presumably involved in metal binding include Asp-8, His-10, Asp-36, Asn-59, Asn-60, His-97, His-120, Thr-121, and His-122 (?). All are present except Thr-121, which is replaced by Ser; however, because it is like Thr, the presence of conserved metal-binding residues suggests possible phosphodiesterase activity; however, the relatively low sequence identity (22.50%) limits confidence in this prediction.
C1KZM7
The protein C1KZM7 comprises 156 residues, and the model was successfully built for residues 3–142. The overall structure of this protein is shown in Figure. Our model shares 30.41% sequence identity with the template and has an RMSD of 0.085 Å. Furthermore, our model contains 5 α-helices and 5 β-sheets. It has been found that the residues Arg135, Ser131, Val38, Pro8, Gly117, Gln119, Ala133, Val132, Gly120, Gly123, and Asn122 are responsible for ATP binding (?). Some residues are conserved in our model, while some are not. We can tentatively conclude that this protein may function as a stress protein and as an ectoine producer, which helps the bacteria to survive osmotic stress. But it cannot be said with certainty. Additionally, during the ProFunc analysis of the model, 1 significant ligand-binding template was identified out of 94569 ligand-binding templates.
C1KYZ6
The protein C1KYZ6 is 362 residues long, and the model was successfully built for residues 42–196. The overall structure of this protein is shown in Figure. Our model shares 12.35% sequence identity with the template and has an RMSD of 5.687 Å. Furthermore, our model contains 4 α-helices and 7 β-sheets. The residues Ile 342, Asn 346, Tyr 465, and Leu 469 are responsible for the formation of the crevice region where many bound water molecules were observed (Xu et al., 2009). This crevice can virtually divide the domain into two parts: the upper and lower subdomains. These are the only residues of importance observed in the template. Due to very low sequence identity (12.35%) and high RMSD (5.687 Å), the structural model lacks sufficient reliability for functional inference.
C1KYT3
The protein C1KYT3 is 211 residues long, and the model was successfully built for residues 5–201. The overall structure of this protein is shown in Figure. Our model shares 33.65% sequence identity with the template and has an RMSD of 3.75 Å. Furthermore, our model contains 5 α-helices and 8 β-sheets. The residues Ser23, His54, Glu77, and Arg104 have been shown to participate in catalysis (?). The residues Lys22, Arg24, Phe25, Asp75, Gly76, Pro78, Ala82, Tyr105, Tyr106, Gly107, Leu111, Leu116, Tyr120, Asp74, and Thr81 probably contribute to substrate binding mechanism of the protein. In the ProFunc analysis, 1 significant DNA-binding template was identified among 4,194 available DNA-binding templates. Because most residues are conserved in our model, the protein's function may involve differentiation, development, and regulation of cell proliferation.
C1KY51
The protein C1KY51 comprises 147 residues, and the model was successfully built for residues 6–145. The overall structure of this protein is shown in Figure. Our model shares 46.00% sequence identity with the template and has an RMSD of 3.3 Å. Furthermore, our model contains 5 α-helices and 3 β-sheets. In the template structure, the zinc-binding region is positioned at the apex of the structural core, which consists of three antiparallel β-strands flanked by two α-helices. This site is exposed on the protein surface and remains accessible to solvent molecules. The coordinated zinc ion interacts with three conserved cysteine residues, Cys40, Cys65, and Cys127, as well as an additional conserved residue, Asp42 (Liu et al., 2005). The geometry of coordination between the zinc ion and the sulfur atoms of the cysteines, along with the OD1 atom of Asp42, is tetrahedral, with an average bond distance of approximately 2.5 Å. Within the conserved PXCGD motif, Cys40 represents one of the essential cysteines required for activity. Asp42 is another invariant residue in this motif and has been shown to significantly stabilize IscU–[Fe2S2]2- complexes in both Thermotoga maritima and human IscU proteins (Liu et al., 2005).
Electron density corresponding to the side chains of Asn36 and Asn37 in Loop 2 is absent, and the density is discontinuous between Pro38 and Thr39. In contrast, Cys40, Gly41, and Asp42 are clearly resolved. Loop 3 comprises Gly64 and Cys65, residues that are highly conserved among members of the IscU/NifU protein families. Cys65 (equivalent to Cys63 in E. coli) is another critical cysteine known to participate in disulfide bond formation with an active-site cysteine of IscS in E. coli. The third essential cysteine, Cys127, is situated on the upper segment of helix α6 and is well defined in the Sp_IscU structure. Arg124, located near the zinc-binding region at the N-terminus of helix α6, is also clearly resolved and highly conserved across IscU/NifU proteins (Liu et al., 2005). Due to its proximity to Cys65, Arg124 may contribute to stabilization of the S2 intermediate during sulfur transfer from IscS to IscU. Surface analysis of the template further revealed a conserved negatively charged region formed by acidic residues Asp17, Asp28, Asp57, Asp77, and Glu56. ProFunc analysis identified one statistically significant ligand-binding template among 94,569 screened entries. Considering the conservation pattern of key residues and structural features, C1KY51 is likely involved in processes such as electron transfer, regulatory mechanisms, environmental sensing, and substrate activation, although experimental validation is necessary to confirm these proposed functions.
C1KYL6
The protein C1KYL6 is 273 residues long, and the model was successfully built for residues 4–271. The overall structure of this protein is shown in Figure. Our model shares 25.19% sequence identity with the template and has an RMSD of 0.091 Å. Furthermore, our model contains 8 α-helices and 12 β-sheets. The residues Arg45, Asp10, Thr43, Asp8, Lys185, Asn211, and Asp208 have been identified to constitute the template's active site (Lu et al., 2011), and these residues are conserved in our protein as well. Hence, the function of our protein can be predicted as the same as that of the HAD superfamily phosphatases, which are involved in phosphoryl transfer reactions and hydrolytic dephosphorylation.
C1KY50
The protein C1KY50 comprises 464 residues, and the model was successfully built for residues 33–462. The overall structure of this protein is shown in Figure. Our model shares 17.20% sequence identity with the template and has an RMSD of 7.538 Å. Furthermore, our model contains 8 α-helices and 18 β-sheets. The residues Phe373, Leu375, Ile380, Met388, Ile389, Ala392, and Ala395 are involved in hydrophobic interactions (Wada et al., 2009). Iron-sulfur (Fe-S) proteins that contain a Fe-S cluster as a prosthetic group, such as our template, are widely utilized in organisms for a variety of cellular processes, including respiratory and photosynthetic electron transport, and in the regulation of gene expression (Wada et al., 2009). Given the low sequence identity (17.20%) and high RMSD (7.538 Å), the structural model of C1KY50 should be interpreted with caution. Although conserved residues suggest a possible iron–sulfur cluster–related role, experimental validation is necessary.
This comprehensive in silico analysis enabled functional annotation of 92 hypothetical proteins from Listeria monocytogenes. Sequence-based classification identified multiple functional classes, while structural modeling provided additional mechanistic insight for selected proteins. Although several predictions were strongly supported by conserved catalytic residues and structural similarity, others require cautious interpretation due to low sequence identity or model reliability. The study improves genome annotation quality and identifies promising candidates for subsequent biochemical and functional validation.
4. Conclusions
We have conducted an extensive analysis of the HPs of Listeria monocytogenes. Following genome analysis of pathogens, we observed numerous proteins whose functions remain uncharacterized. Comparative genomics indicates that these proteins are present across organisms but have not yet been functionally characterized to date. The first part of this study comprises proteins for which biochemical activity can be predicted with reasonable confidence using genomic sequence-based approaches. We listed the proposed functions of most HPs using sequence analysis tools. The second part includes structure determination and the development of a structure-function relationship. We have successfully determined the structure-function relationship for 13 HPs from Listeria monocytogenes. This structural genomics project enables us to better understanding of pathogenesis of this microorganism. We have finally achieved our goal of assigning structure-based biochemical functions to most hypothetical proteins from Listeria monocytogenes. However, further biochemical work is needed to assign functions with accurate precision.
5. Conflict of Interest:
There is no conflict of interest to declare.
6. Data Availability Statement:
The data supporting this study are provided in this article.
7. Funding:
None
8. Acknowledgements:
None
9. Supplementary materials:
None
Figures
Figure 1. Outline of structure and function assignment to the hypothetical proteins.
{kind=link}
Figure 2. Structural superimposition of modeled proteins C1L1U8, C1KYM2, C1KYY1, C1L0M8, C1L0R8, and C1L165 with their respective template structures. Cartoon representations showing the three-dimensional models superimposed over their corresponding template structures.
{kind=link}
Figure 3. Structural superimposition of modeled proteins C1L2E5–C1KY50 with their respective template structures. Cartoon representations depicting the structural alignment of modeled proteins with their corresponding templates.
{kind=link}
References
- 1
Adams, P.D., Afonine, P.V., Bunkoczi, G., et al. 2010, Acta Crystallogr. D Biol. Crystallogr., 66, 213–221
- 2
Agustiandari, H, Lubelski, J, van, den Berg van Saparoea HB, et al. 2008, J Bacteriol, 190, 759–763
- 3
Aravind, L., Koonin, E., V 1998, Sci., 23, 469–472
- 4
Brunger, & A.T 1996, X-PLOR, Version 3.85, (New Haven, CT: Yale University Press)
- 5
Britton, R.A., Wen, T., Schaefer, L., et al. 2007, Mol. Microbiol., 63, 127–138
- 6
Bugrysheva, J.V., Scott, J.R 2010, Mol. Microbiol., 75, 731–743
- 7
Carballido-Lopez, R, Formstone, A, Li, Y, et al. 2006, Dev Cell, 11, 399–409
- 8
Carniol, K, Ben-Yehuda, S, King, N, Losick, R 2005, J Bacteriol, 187, 3511–3520
- 9
Cheung, HY, & Freese, E 1985, J Bacteriol, 161, 1222–1225
- 10
Chin, Ko-Hsin, Tsai, et al. 2007, The crystal structure of XC1258 from Xanthomonas campestris: A putative procaryotic Nit protein with an arsenic adduct in the active site
- 11
Claessen, D, Emmins, R, Hamoen, LW, et al. 2008, Mol Microbiol, 68, 1029–1046
- 12
Collins, M.D., & Jones, D 1981, Microbiol. Rev., 45, 316–354
- 13
Datta, S., Mori, Y., Takagi, K., et al. 2001, Proc. Natl. Acad. Sci., 98, 14268–14273
- 14
Eva, S. Schweikhard, Sonja, I. Kuhlmann, Hans-Jörg, Kunte, et al. 2010, Biochemistry, 49, 2194–2204
- 15
Fibriansah, G, Kovacs, AT, Pool, TJ, et al. 2012, PLoS ONE, 7, e48015
- 16
Finn, R., D., Tate, et al. 2008, Nucleic Acids Res
- 17
Flower, & D.R 1996, Biochem J., 318, 1–14
- 18
Frances, Park, Ketan, Gajiwala, Galina, Eroshkina, et al. 2004, Proteins, 55, 775–777
- 19
Gury, J, Barthelmebs, L, Tran, NP, et al. 2004, Appl Environ Microbiol, 70, 2146–2153
- 20
Handa, N., Terada, T., Doi-Katayama, Y., et al. 2004, Protein Sci., 14, 1004–1010
- 21
Huber, H., E., Beauchamp, et al. 1988, Biol. Chem., 263, 13549–13556
- 22
Ivan, I. Vorontsov, George, Minasov, Olga, Kiryukhina, et al. 2011, J Biol Chem, 286, 33158–33166
- 23
Jones, T.A., Zou, J.Y., Cowan, S.W., Kjeldgaard, M 1991, Acta Crystallogr. A, 47, 110–119
- 24
Joseph, A. Newman, Lorraine, Hewitt, Cecilia, Rodrigues, et al. 2010, Unusual, Dual Endo- and Exonuclease Activity in the Degradosome Explained by Crystal Structure Analysis of RNase J1
- 25
Kathrin, Schirner, Jon, Marles-Wright, Richard, J Lewis, Jeff, Errington 2009, EMBO J., 28, 830–842
- 26
Kornberg, S., R., Lehman, et al. 1958, Biol. Chem., 233, 159–162
- 27
Kovacikova, G, Lin, W, Skorupski, K 2003, J Bacteriol, 185, 4825–4836
- 28
Kei, Wada, Norika, Sumi, Rina, Nagai, et al. 2009, J Mol Biol, 387, 245–258
- 29
Liu, Jinyu, Oganesyan, et al. 2005, Proteins, 59, 875–881
- 30
Lu, Zhibing, Dunaway-Mariano, et al. 2011, thetaiotaomicron. Proteins, 79, 3099–3107
- 31
Mengaud, J., Geoffroy, C., Cossart, P 1991, Infect. Immun., 59, 1043–1049
- 32
Mengaud, J., Vicente, et al. 1988, Infect. Immun., 56, 766–772
- 33
Mengaud, J., Vicente, M.-F., Cossart, P 1989, Infect. Immun., 57, 3695–3701
- 34
Michard, J., Jardy, N., Laine, K 1989, Microbiol. Aliments Nutr., 7, 31–42
- 35
Miller, J., K., Burns, J 1970, Appl. Microbiol., 19, 772–775
- 36
Morris, I., J., Ribeiro, C., D 1989, Lancet, ii, 1285–1286
- 37
Mulder, C., J. J., Zanen, H., C 1986, Eur. J. Pediatr., 145, 60–62
- 38
Munk, M., E., Kaufmann, S., H. E 1988, Microb. Pathog., 5, 49–54
- 39
Murray, E., G. D., Webb, et al. 1926, sp.). J. Pathol. Bacteriol., 29, 407–439
- 40
Newman, Joseph, A., Hewitt, et al. 2010, Unusual, Dual Endo- and Exonuclease Activity in the Degradosome Explained by Crystal Structure Analysis of RNase J1
- 41
Nicolai-Scholten, M.-E., Potel, J., et al. 1985, Immun. Infekt., 13, 76–77
- 42
Nicolas, J.-A., Vidaud, N 1987, Rec. Med. Vet., 163, 283–285
- 43
Nieman, R., E., Lorber, B 1980, Rev. Infect. Dis., 2, 207–227
- 44
Njoku-Obi, A., N., Jenkins, et al. 1963, J. Bacteriol., 86, 1–8
- 45
Nocera, D., Bannerman, E., Rocourt, J., et al. 1990, J. Clin. Microbiol., 28, 2259–2263
- 46
Northolt, M., D., Beckers, et al. 1988, Neth. Milk Dairy J., 42, 207–219
- 47
Notermans, S., Chakraborty, T., Leimeister-Wachter, M., et al. 1989, Appl. Environ. Microbiol., 55, 902–906
- 48
Olander, A., Harlander, S., Swaminathan, B 1990, Abstr
- 49
Olsen, J., A., Yousef, et al. 1988, Milchwissenschaft, 43, 487–498
- 50
Ortel, S 1968, Dtsch. Gesundheitswes., 23, 753–759
- 51
Ortel, S 1975, Zentralbl. Bakteriol. Mikrobiol. Hyg. I. Abt. Orig. A, 231, 491–502
- 52
Ortel, S 1989, Acta Microbiol. Hung., 36, 153–157
- 53
Palumbo, S., A., Williams, A., C 1990, J. Food Prot., 53, 377–381
- 54
Papageorgiou, D., K., Marth, E., H 1989, J. Food Prot., 52, 459–465
- 55
Parish, M., E., Higgins, D., P 1989, J. Food Prot., 52, 144–147
- 56
Parodi, M., Marino, P., Balzaretti, C., et al. 1990, G. Mal. Infett. Parassit., 42, 115–116
- 57
Parrisius, J., Bhakdi, S., Roth, M., et al. 1986, Infect. Immun., 51, 314–319
- 58
Paterson, & J., S 1940, J. Pathol. Bacteriol., 51, 427–436
- 59
Pearson, L., J., Marth, E., H 1990, J. Food Prot., 53, 30–37
- 60
Peel, M., Donachie, W., Shaw, A 1988, J. Gen. Microbiol., 143, 2171–2178
- 61
Peeters, A., Waer, M., Michielsen, P., et al. 1989, Clin. Neurol. Neurosurg., 91, 29–36
- 62
Perez-Diaz, J., C., Vicente, et al. 1982, Plasmid, 8, 112–118
- 63
Peterkin, P., I., Idziak, et al. 1989, Food Microbiol., 6, 281–284
- 64
Peterkin, P., I., Idziak, et al. 1991, Appl. Environ. Microbiol., 57, 586–591
- 65
Piffaretti, J.-C., Kressbuch, et al. 1989, Proc. Natl. Acad. Sci. USA, 86, 3818–3822
- 66
Pine, L., Malcolm, et al. 1989, Can. J. Microbiol., 35, 245–254
- 67
Pine, L., Malcolm, et al. 1990, Infect. Immun., 58, 2940–2945
- 68
Pini, P., N., Gilbert, R., J 1988, Int. J. Food Microbiol., 7, 331–337
- 69
Pollock, S., S., Pollock, et al. 1984, Q. J. Med., LIII, 331–340
- 70
Portnoy, D., A., Jacks, et al. 1988, J. Exp. Med., 167, 1459–1471
- 71
Poyart-Salmeron, C., Carlier, C., Trieu-Cuot, P., et al. 1990, Lancet, 335, 1422–1426
- 72
Pritchard, T., J., Donnelly, C., W 1990, Abstr
- 73
Racz, P., Tenner, K., Mero, E 1972, Lab. Invest., 26, 694–700
- 74
Ralovich, B 1984, Listeriosis research, present situation and perspective
- 75
Ramsden, G., H., Johnson, et al. 1989, Listeriosis in immunocompromised pregnancy
- 76
Rao, N 1989, J. Thorac. Cardiovasc. Surg., 98, 303–304
- 77
Reetz, J., Schilow, W., F., Schwebs, M 1989, Occurrence of Listeria monocytogenes in cell nuclei
- 78
Riedo, F., X., Pinner, et al. 1990, Program Abstr
- 79
Rocourt, J 1988, Taxonomy of the genus Listeria
- 80
Rocourt, J., Espaze, et al. 1989, Lancet, ii, 1217–1218
- 81
Rocourt, J., Audurier, et al. 1985, Zentralbl. Bakteriol., 259, 489–497
- 82
Rocourt, J., Grimont, et al. 1982, Curr. Microbiol., 7, 383–388
- 83
Rocourt, J., Wehmeyer, U., Stackebrandt, E 1987, nov. Int. J. Syst. Bacteriol., 37, 266–270
- 84
Rodler, M., & Korbler, W 1989, Acta Microbiol. Hung., 36, 259–261
- 85
Rodriguez, L., D., Boland, et al. 1986, J. Clin. Microbiol., 24, 99–103
- 86
Roncoroni, A., J., Michans, et al. 1987, Medicina (Buenos Aires), 47, 239–242
- 87
Rosenow, E., M., Marth, E., H 1987, J. Food Prot., 50, 452–459
- 88
Ryser, E., T., Marth, E., H 1987, J. Food Prot., 50, 7–13
- 89
Ryser, E., T., Marth, E., H 1988, Can. J. Microbiol., 34, 730–734
- 90
Shengfeng, Chen, Yakunin, A., F., et al. 2004, J. Biol. Chem., 279, 31854–31862
- 91
Takai, N., Ikeuchi, S., Manabe, K., Kutsuna, S 2006, J. Biol. Rhythms, 21, 235–244
- 92
Tran, N., P., Gury, et al. 2008, J. Bacteriol., 190, 3213–3224
- 93
Vorontsov, I., I., Minasov, et al. 2011, J. Biol. Chem., 286, 33158–33166
- 94
Wada, K., Sumi, N., Nagai, R., et al. 2009, J. Mol. Biol., 387, 245–258
- 95
Xu, Yongbin, Sim, et al. 2009, Biochemistry, 48, 5218–5225
Tables
Table 1. List of hypothetical proteins in Listeria monocytogenes (Part 1 of 3).
| p0.19 p0.06 p0.07 p0.20 p0.34 S. No. | Name of Hypothetical Protein | pI | Length | Pfam | Proposed Function |
|---|---|---|---|---|---|
| 1 | C1L300/ C1L300_LISMC | 9.69 | 447 | MatE (PF01554) | antiporter activity, drug transmembrane transporter activity |
| 2 | C1L2X5/ C1L2X5_LISMC | 7.85 | 345 | UPF0118 (PF01594) | NA |
| 3 | C1L2V8/ C1L2V8_LISMC | 5.02 | 120 | DUF964 (PF06133) | NA |
| 4 | C1L2V7/ C1L2V7_LISMC | 5.45 | 267 | YmdB (PF13277) | putative phosphoesterases |
| 5 | C1L2S2/ C1L2S2_LISMC | 6.41 | 274 | FtsJ (PF01728), S4 (PF01479) | methyltransferase involved in viral RNA capping, RNA binding |
| 6 | C1L2Q9/ C1L2Q9_LISMC | 6.47 | 321 | DUF1385 (PF07136) | NA |
| 7 | C1L2P0/ C1L2P0_LISMC | 4.78 | 118 | No domains | NA |
| 8 | C1L2N2/ C1L2N2_LISMC | 5.44 | 92 | DUF503 (PF04456) | NA |
| 9 | C1L2K8/ C1L2K8_LISMC | 5.38 | 174 | Acetyltransf_3 (PF13302) | transfers acetyl group |
| 10 | C1L2J0/ C1L2J0_LISMC | 7.84 | 103 | NA | NA |
| 11 | C1L2E5/ C1L2E5_LISMC | 5.13 | 174 | Metallophos_2 (PF12850) | Phosphoesterase (hydrolase activity, acting on ester bonds) |
| 12 | C1L1Z7/ C1L1Z7_LISMC | 6.42 | 774 | Glycos_transf_2 (PF00535), Glyphos_transf (PF04464) | glycerophosphotransferase (sequential transfer of glycerol-phosphate units) (CDP-glycerol glycerophosphotransferase activity) |
| 13 | C1L1V8/ C1L1V8_LISMC | 9.43 | 267 | DUF817 (PF05675) | NA |
| 14 | C1L1U8/ C1L1U8_LISMC | 6.18 | 555 | Lactamase_B (PF00753), RMMBL_DRMBL (CL0398) | RNA binding, hydrolase activity, acting on ester bonds, metal ion binding |
| 15 | C1L1S5/ C1L1S5_LISMC | 10.1 | 344 | DUF218 (PF02698) | NA |
| 16 | C1L1R3/ C1L1R3_LISMC | 9.3 | 255 | TerC (PF03741) | integral to membrane |
| 17 | C1L1R2/ C1L1R2_LISMC | 9.36 | 255 | TerC (PF03741) | integral to membrane |
| 18 | C1L1R1/ C1L1R1_LISMC | 9.09 | 453 | MatE (PF01554) | antiporter activity, drug transmembrane transporter activity |
| 19 | C1L1K4/ C1L1K4_LISMC | 9.81 | 201 | SNARE_assoc (PF09335) | NA |
| 20 | C1L1G8/ C1L1G8_LISMC | 5.59 | 725 | Tex_N (PF09371), HHH_3 (PF12836), S1 (PF00575) | RNA binding, hydrolase activity- acting on ester bonds |
| 21 | C1L1B3/ C1L1B3_LISMC | 4.78 | 125 | Ribonuc_L-PSP (PF01042) | Inhibits protein synthesis by cleaving the mRNA |
| 22 | C1L190/ C1L190_LISMC | 5.09 | 282 | NA | NA |
| 23 | C1L165/ C1L165_LISMC | 4.69 | 176 | YceI (PF04264) | binds to polyisoprenoid |
| 24 | C1L164/ C1L164_LISMC | 9.54 | 306 | EamA (PF00892) | transporter activity |
| 25 | C1L162/ C1L162_LISMC | 9.27 | 233 | DUF554 (PF04474) | NA |
| 26 | C1L161/ C1L161_LISMC | 4.79 | 296 | CN_hydrolase (PF00795) | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds |
| 27 | C1L158/ C1L158_LISMC | 5.68 | 282 | PhzC-PhzF (PF02567) | Phenazine biosynthes |
| 28 | C1L143/ C1L143_LISMC | 5.33 | 309 | DAGK_cat (PF00781) | NAD+ kinase activity, diacylglycerol kinase activity |
| 29 | C1L0U3/ C1L0U3_LISMC | 4.83 | 288 | Hydrolase_3 (PF08282) | hydrolase activity |
| 30 | C1L0T5/ C1L0T5_LISMC | 5.42 | 209 | HhH-GPD (PF00730) | DNA binding, endonuclease activity |
| 31 | C1L0T0/ C1L0T0_LISMC | 9.82 | 187 | DUF420 (PF04238) | NA |
Table 2. List of hypothetical proteins in Listeria monocytogenes (Part 2 of 3).
| p0.19 p0.06 p0.07 p0.20 p0.34 S. No. | Name of Hypothetical Protein | pI | Length | Pfam | Proposed Function |
|---|---|---|---|---|---|
| 32 | C1L0R8/ C1L0R8_LISMC | 5.34 | 606 | Sulfatase (PF00884) | sulfuric ester hydrolase activity |
| 33 | C1L0Q0/ C1L0Q0_LISMC | 9.71 | 246 | TauE (PF01925) | NA |
| 34 | C1L0P3/ C1L0P3_LISMC | 4.87 | 172 | Acetyltransf_1 (PF00583) | acetyltransferase activity |
| 35 | C1L0M8/ C1L0M8_LISMC | 4.78 | 110 | PadR (PF03551) | involved in negative regulation of phenolic acid metabolism |
| 36 | C1L0L0/ C1L0L0_LISMC | 6.08 | 394 | Methyltrans_SAM (PF10672) | methyltransferase activity |
| 37 | C1L0K2/ C1L0K2_LISMC | 7.7 | 431 | Xan_ur_permease (PF00860) | transporter activity |
| 38 | C1L0G9/ C1L0G9_LISMC | 4.82 | 264 | AP_endonuc_2 (PF01261) | involved in the myo-inositol catabolism (isomerase activity) |
| 39 | C1L075/ C1L075_LISMC | 5.18 | 346 | Lactonase (PF10282) | 6-phosphogluconolactonase activity |
| 40 | C1L028/ C1L028_LISMC | 5.72 | 143 | Usp (PF00582) | response to stress |
| 41 | C1L024/ C1L024_LISMC | 5.26 | 226 | GATase (PF00117) | transferase activity |
| 42 | C1KZV6/ C1KZV6_LISMC | 4.89 | 281 | NmrA (PF05368) | nucleotide binding (negative transcriptional regulator involved in the post-translational modification of the transcription factor AreA.) |
| 43 | C1KZU1/ C1KZU1_LISMC | 5.41 | 377 | Gly_kinase (PF02595) | glycerate kinase activity |
| 44 | C1KZQ3/ C1KZQ3_LISMC | 4.64 | 279 | Hydrolase_3 (PF08282) | hydrolase activity |
| 45 | C1KZM7/ C1KZM7_LISMC | 8.93 | 156 | Usp (PF00582) | response to stress |
| 46 | C1KZM4/ C1KZM4_LISMC | 6.15 | 120 | YjbR (PF04237) | NA |
| 47 | C1KZE1/ C1KZE1_LISMC | 4.83 | 270 | Hydrolase_3 (PF08282) | hydrolase activity |
| 48 | C1KZ86/ C1KZ86_LISMC | 5.18 | 111 | PhnA_Zn_Ribbon (PF08274), PhnA (PF03831) | NA |
| 49 | C1KZ81/ C1KZ81_LISMC | 5.97 | 494 | FTR1 (PF03239) | transmembrane transport |
| 50 | C1KZ17/ C1KZ17_LISMC | 8.78 | 220 | MgtC (PF02308) | transport of Mg2+ |
| 51 | C1KZ02/ C1KZ02_LISMC | 5.31 | 280 | SAM_adeno_trans (PF01887) | NA |
| 52 | C1KYZ6/ C1KYZ6_LISMC | 8.7 | 362 | MacB_PCD (PF12704), FtsX (PF02687) | transport lipids targeted to the outer membrane across the inner membrane |
| 53 | C1KYZ4/ C1KYZ4_LISMC | 5.44 | 97 | ABM (PF03992) | monooxygenase activity |
| 54 | C1KYZ2/ C1KYZ2_LISMC | 4.62 | 275 | Hydrolase_3 (PF08282) | hydrolase activity |
| 55 | C1KYY1/ C1KYY1_LISMC | 5.4 | 440 | HD (PF01966) | metal ion binding, phosphoric diester hydrolase activity |
| 56 | C1KYX9/ C1KYX9_LISMC | 6.83 | 217 | Peptidase_M50 (PF02163) | proteolysis, metalloendopeptidase activity |
| 57 | C1KYX3/ C1KYX3_LISMC | 9.03 | 306 | DAGK_cat (PF00781) | kinase, transferase |
| 58 | C1KYW9/ C1KYW9_LISMC | 10.1 | 357 | UPF0104 (PF03706) | NA |
| 59 | C1KYT3/ C1KYT3_LISMC | 5.78 | 211 | UPF0029 (PF01205), DUF1949 (PF09186) | GTP binding |
| 60 | C1KYS5/ C1KYS5_LISMC | 6.43 | 287 | DUF161 (PF02588), DUF2179 (PF10035) | NA |
| 61 | C1KYP0/ C1KYP0_LISMC | 4.97 | 322 | UPF0052 (PF01933) | NA |
| 62 | C1KYM2/ C1KYM2_LISMC | 5.12 | 259 | CN_hydrolase (PF00795) | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds |
Table 3. List of hypothetical proteins in Listeria monocytogenes (Part 3 of 3).
| p0.19 p0.06 p0.07 p0.20 p0.34 S. No. | Name of Hypothetical Protein | pI | Length | Pfam | Proposed Function |
|---|---|---|---|---|---|
| 63 | C1KYL6/ C1KYL6_LISMC | 5.39 | 273 | Hydrolase_3 (PF08282) | ATP binding, ATPase activity, coupled to transmembrane movement of ions, phosphorylative mechanism |
| 64 | C1KYI4/ C1KYI4_LISMC | 5.98 | 201 | MTS (PF05175) | 16S rRNA (guanine(966)-N(2))-methyltransferase activity |
| 65 | C1KY64/ C1KY64_LISMC | 5.76 | 117 | ArsC (PF03960) | regulate the transcription of multiple genes in response to disulfide stress |
| 66 | C1KY61/ C1KY61_LISMC | 5.27 | 291 | Cation_efflux (PF01545) | cation transmembrane transporter activity |
| 67 | C1KY51/ C1KY51_LISMC | 5.94 | 147 | NifU_N (PF01592) | iron ion binding, iron-sulfur cluster binding |
| 68 | C1KY50/ C1KY50_LISMC | 4.87 | 464 | UPF0051 (PF01458) | iron-sulfur cluster assembly |
| 69 | C1KY46/ C1KY46_LISMC | 9.02 | 279 | TauE (PF01925) | involved in the transport of anions across the cytoplasmic membrane during taurine metabolism as an exporter of sulfoacetate |
| 70 | C1KY45/ C1KY45_LISMC | 5.08 | 462 | Metallophos (PF00149), 5_nucleotid_C (PF02872) | hydrolase activity (5_nucleotid_C) |
| 71 | C1KY43/ C1KY43_LISMC | 4.94 | 255 | Hydrolase_like (PF13242), Hydrolase_6 (PF13344) | hydrolase activity |
| 72 | C1KY41/ C1KY41_LISMC | 4.82 | 436 | DUF21 (PF01595), CBS (PF00571), CorC_HlyC (PF03471) | flavin adenine dinucleotide binding (CBS) |
| 73 | C1KY30/ C1KY30_LISMC | 9.43 | 406 | Voltage_CLC (PF00654) | voltage-gated chloride channel activity |
| 74 | C1KY03/ C1KY03_LISMC | 5.29 | 156 | Rrf2 (PF02082) | NA |
| 75 | C1KXZ5/ C1KXZ5_LISMC | 4.65 | 281 | Hydrolase_3 (PF08282) | hydrolase activity |
| 76 | C1KXZ0/ C1KXZ0_LISMC | 4.76 | 532 | Amidohydro_3 (PF07969) | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in cyclic amides |
| 77 | C1KXY4/ C1KXY4_LISMC | 6.26 | 257 | Methyltransf_26 (PF13659) | methyltransferase activity |
| 78 | C1KXX7/ C1KXX7_LISMC | 4.83 | 270 | Hydrolase_3 (PF08282) | hydrolase activity |
| 79 | C1KXV5/ C1KXV5_LISMC | 5.52 | 249 | EAL (PF00563) | NA |
| 80 | C1KXN7/ C1KXN7_LISMC | 6.53 | 331 | Bac_luciferase (PF00296) | oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen |
| 81 | C1KXN1/ C1KXN1_LISMC | 6.58 | 147 | DUF523 (PF04463) | NA |
| 82 | C1KWJ0/ C1KWJ0_LISMC | 6.74 | 121 | DUF1798 (PF08807) | NA |
| 83 | C1KWG8/ C1KWG8_LISMC | 6.52 | 327 | DUF939 (PF06081), DUF939_C (PF11728) | NA |
| 84 | C1KWG7/ C1KWG7_LISMC | 4.39 | 125 | Glyoxalase_2 (PF12681) | NA |
| 85 | C1KWG4/ C1KWG4_LISMC | 6.52 | 205 | HTH_11 (PF08279), CBS (PF00571) | sequence-specific DNA binding transcription factor activity (CBS) |
| 86 | C1KWC7/ C1KWC7_LISMC | 5.29 | 291 | YicC_N (PF03755), DUF1732 (PF08340), DUF1732 (PF08340) | play a role in the stationary phase survival (YicC) |
| 87 | C1KW03/ C1KW03_LISMC | 6.11 | 126 | OsmC (PF02566) | has a novel pattern of oxidative stress regulation |
| 88 | C1KVW1/ C1KVW1_LISMC | 6.22 | 192 | rRNA_methylase (PF06962) | methyltransferase activity |
| 89 | C1KVW0/ C1KVW0_LISMC | 5.48 | 321 | Radical_SAM (PF04055) | catalytic activity, iron-sulfur cluster binding |
| 90 | C1KVA0/ C1KVA0_LISMC | 6.02 | 234 | DUF633 (PF04816) | tRNA (adenine-N1-)-methyltransferase activity |
| 91 | C1KV99/ C1KV99_LISMC | 5.5 | 373 | NIF3 (PF01784) | NIF3 interacts with the yeast transcriptional coactivator NGG1p, which is part of the ADA complex |
| 92 | C1KV71/ C1KV71_LISMC | 4.92 | 269 | Hydrolase_3 (PF08282) | cation transport, ATP binding, ATPase activity, coupled to transmembrane movement of ions, phosphorylative mechanism |
Table 4. Major Classes of Hypothetical Proteins in Listeria monocytogenes
| p4.2cm|p9.5cm| S. No. | Proposed function of Hypothetical protein | Accession number |
|---|---|---|
| 1 | Hydrolase | C1L0U3, C1L0R8, C1KZQ3, C1KZE1, C1KYZ2, C1KYY1, C1KY45, C1KY43, C1KXZ5 |
| 2 | Transferase | C1L2K8, C1L1Z7, C1L0P3, C1L0L0, C1L024, C1KYI4, C1KXY4, C1KVW1, C1KVA0 |
| 3 | Transporter | C1L300, C1L1R1, C1L164, C1L0K2, C1KZ81, C1KZ17, C1KYZ6, C1KY61, C1KY46 |
| 4 | RNA binding | C1L2S2, C1L1U8, C1L1G8 |
| 5 | Hydrolase acting on carbon-nitrogen but not on peptide | C1L161, C1KYM2, C1KXZ0 |
| 6 | Kinase | C1L143, C1KZU1, C1KYX3 |
| 7 | Response to stress | C1LO28, C1KZM7, C1KW03 |
| 8 | Integral membrane | C1L1R3, C1L1R2, C1KY64 |
| 9 | DNA binding | C1L0T5, C1KWG4 |
| 10 | Phosphoesterase | C1L2V7, C1L2E5 |
| 11 | ATP Binding | C1KYL6, C1KV71 |
| 12 | Others | C1L1B3, C1L165, C1L158, C1L0M8, C1L0G9, C1L075, C1KZV6, C1KYZ4, C1KYX9, C1KYT3, C1KY51, C1KY50, C1KY41, C1KY30, C1KXN7, C1KWC7, C1KVW8, C1KV99 |
| 13 | Unknown | C1L2X5, C1L2V8, C1L2Q9, C1L2P0, C1L2J0, C1L1S5, C1L1K4, C1L198, C1L162, C1L0T0, C1L0Q0, C1KZM4, C1KZ86, C1KZ02, C1KYW9, C1KYS5, C1KYP0, C1KY03, C1KYN7, C1KXN1, C1KWJ0, C1KWG8, C1KWG7 |
Table 5. Superimposed residues between template and modeled proteins C1L1U8–C1L165. Residue pairs identified through structural superimposition of each modeled protein with its corresponding template structure. Amino acid residues from the template are shown on the left, followed by the aligned residues in the model (Template → Model). Identical residue matches and positional shifts are indicated according to their residue number in the respective structures.
| |p0.80| Protein ID | Superimposed Residues (Template → Model) |
|---|---|
| C1L1U8 | Asp78→Asp78, His79→His79, Asp164→Asp164, His390→His390, His74→His74, His76→His76, His142→His142, Asp195→Asp195, His368→His368, Gly49→Gly49, Asp51→Asp51, Asp443→Asp443, Glu464→Glu464, Asp449→Asp449, Arg544→Arg544, Ser366→Ser366, His364→His364, Gly367→Gly367, Ser233→Ser233, Glu77→Glu77, Phe42→Phe42, Tyr52→Tyr52 |
| C1KYM2 | Glu43→Glu42, Lys109→Lys111, Cys143→Cys145, Tyr144→Tyr146, Phe49→Tyr48, Phe113→Phe115, Trp175→Trp171, Val142→Ile144, Pro176→Pro172 |
| C1KYY1 | His129→His120, Glu122→Glu113, His66→His64, His110→His110, Asp111→Asp102, Asp183→Asp173, Lys14→Lys12, Asn36→Ala34, Gln41→Gln39, Arg326→Arg317, Lys330→Arg321, His114→His105, Tyr239→Tyr231, Arg63→Arg61, Leu49→Leu47, His119→His110, Tyr187→Tyr177, Tyr243→Tyr235, Tyr368→Tyr358 |
| C1L0M8 | Gly25→Gly27, Tyr26→Tyr28, Glu42→Glu42, Arg71→Arg71, Lys72→Lys72, Tyr46→Tyr46, Arg51→Arg51 |
| C1L0R8 | Thr297→Thr272, His412→His388, Glu253→Glu230, Trp350→Tyr325, His472→His449, Asp471→Asp448, His343→His318, Asn345→Asn320, Arg352→Arg327, Thr408→Ser384 |
| C1L165 | His18→His21, Arg62→Arg66, His65→His69, Trp146→Tyr151 |
Table 6. Superimposed residues between template and modeled proteins C1L2E5–C1KY50. Residue correspondences obtained from structural alignment between each template and its respective modeled protein. Residues are presented as Template → Model to indicate positional and amino acid conservation or substitution observed upon superimposition.
| |p0.80| Protein ID | Superimposed Residues (Template → Model) |
|---|---|
| C1L2E5 | Asp8→Asp8, His10→His10, Asp36→Ser36, Asn59→Asn54, Asn60→Cys55, His97→His78, His120→His107, Thr121→Ser108, His122→His109 |
| C1KZM7 | Arg135→Tyr130, Ser131→Ser126, Val38→Val40, Pro8→Gly10, Gly117→Gly112, Gln119→Thr114, Ala133→Ser128, Val132→Val127, Gly120→Gly115, Gly123→Ala118, Asn122→Ser117 |
| C1KYZ6 | Leu469→Lys186, Tyr465→Trp182, Asn346→Ala79, Ile342→Thr75 |
| C1KYT3 | Ser23→Ser21, His54→His52, Glu77→Glu74, Arg104→Arg100, Lys22→Lys20, Arg24→Arg22, Phe25→Phe23, Asp75→Asp71, Gly76→Gly72, Pro78→Pro74, Ala82→Ala78, Tyr105→Tyr101, Tyr106→Phe102, Gly107→Gly103, Leu111→Leu107, Leu116→Leu112, Tyr120→Tyr116, Asp74→Asp70, Thr81→Thr77 |
| C1KY51 | Cys40→Cys41, Asp42→Asp43, Cys65→Cys66, Arg124→Arg125, Cys127→Cys128, Gly64→Gly65, Asp57→Ala58, Asp77→Gln78, Glu56→Val57 |
| C1KYL6 | Trp171→Ser187, Phe175→Asn191, Arg45→Arg46, Asp1→Asp12, Thr43→Thr44, Asp8→Asp10, Lys185→Lys201, Asn211→Asn227, Asp208→Asp22 |
| C1KY50 | Phe373→Phe415, Leu375→Leu417, Ile380→Leu422, Met338→Met430, Ile389→Ile431, Ala392→Gly434, Ala395→Glu437 |