


About Article
Identification of Potent Inhibitors of Ephrin type-A receptor 1 using Virtual Screening and Molecular Dynamics Simulation Approach
Abstract
Ephrin type-A receptor 1 (EphA1) is a receptor tyrosine kinase implicated in tumor progression, angiogenesis, metastasis, and immune regulation, making it an attractive therapeutic target in cancer. In the present study, a structure-based virtual screening approach was employed to identify potential natural inhibitors of EphA1. A library of 90,000 natural compounds retrieved from the ZINC database was filtered using Lipinski’s rule of five, yielding 32,901 drug-like molecules for docking analysis. The three-dimensional structure of EphA1 was obtained from the AlphaFold database, and molecular docking was performed using InstaDock. Based on binding affinity scores (-11.2 to -9.4 kcal/mol), the top 320 compounds were shortlisted and further subjected to PAINS filtering, physicochemical evaluation, ADMET prediction, carcinogenicity assessment, and PASS analysis. Two compounds, ZINC12660859 and ZINC12661003, demonstrated favourable drug-likeness, high gastrointestinal absorption, non-carcinogenicity, and predicted antineoplastic activity. Interaction analysis revealed that these compounds bind within the ATP-binding pocket of the kinase domain, forming key interactions with residues such as Lys656 and Asp749. Molecular dynamics simulations over 100 ns of the EphA1–ZINC12660859 complex confirmed structural stability, with stable RMSD (0.26 nm), consistent radius of gyration (1.93 nm), minimal SASA variation, and sustained hydrogen bonding throughout the simulation. Overall, ZINC12660859 exhibited strong binding affinity and dynamic stability, suggesting its potential as a lead candidate for EphA1-targeted anticancer therapeutics.
Keywords
1. Introduction
Receptor tyrosine kinases (RTKs) are critical regulators of cellular communication and play essential roles in cell proliferation, differentiation, migration, and survival. Among RTKs, the Eph receptor family is the largest subgroup and is subdivided into EphA and EphB classes based on ligand specificity and sequence homology (Wu et al., 2022). Eph receptors interact with membrane-bound ephrin ligands to initiate bidirectional signaling pathways that regulate tissue patterning, angiogenesis, immune responses, and neuronal development. Ephrin type-A receptor 1 (EphA1), encoded by the EPHA1 gene located on chromosome 7q34, was first identified in hepatocellular carcinoma cells (Wu et al., 2022). EphA1 plays diverse roles in fundamental cellular processes such as cell movement and attachment (Yamazaki et al., 2009), regulation of immune response and inflammation (Hjorthaug & Aasheim, 2007), cancer progression, nervous system development, and angiogenesis (Wu et al., 2022). During development, EphA1 is involved in blood vessel remodelling, axon guidance, the spatial organisation of diverse cell populations, and the creation of synaptic connections between neurons (Lisabeth et al., 2013). Ephrin-A1 promotes directional mobility of CD8+CCR7+ T lymphocytes, a type of T cell implicated in immunological responses, and thus regulates immunological processes such as lymph node homing and antigen presentation (Hjorthaug & Aasheim, 2007). EphA1–ILK signalling regulates cell adhesion dynamics and cytoskeletal organisation, both of which are required for cellular functions such as migration and tissue formation (Yamazaki et al., 2009).
EphA1 increases cell division and protects cells in bovine mammary epithelial cells by reducing ER stress, inhibiting inflammatory responses, and minimizing cellular damage caused by inflammation (Kang et al., 2018). EphA1 also contributes to neuroinflammation through the CXCL12/CXCR4 signalling pathway and is considered an important drug target for Parkinson's disease (Ma et al., 2021) and Alzheimer's disease (Villegas-Llerena et al., 2016). EphA1 is differentially expressed in different types of cancer and is therefore associated with tumour growth, tumour angiogenesis, invasion, prognosis, and metastasis (Ieguchi & Maru, 2019). EphA1 is overexpressed in thymoma (Yu et al., 2019), prostate cancer (Peng et al., 2013), hepatocellular carcinoma (Wang et al., 2016), ovarian cancer (Adu-Gyamfi et al., 2021), breast cancer (Liang et al., 2021), clear cell renal cell carcinoma (Toma et al., 2014), gastric carcinoma (Wang et al., 2020), nasopharyngeal carcinoma (Dai & Zhang, 2020), and oesophageal squamous cell carcinoma (Wang et al., 2013), while it is downregulated in colorectal cancer (Wu et al., 2016) and uveal melanoma (Gajdzis et al., 2020).
EphA1 has diverse functions across diseases, providing important insights into underlying mechanisms and revealing potential therapeutic targets. EphA1 has been linked to tumour development and metastasis by promoting angiogenesis, cell migration, and epithelial–mesenchymal transition (Ieguchi & Maru, 2019). EphA1 has also been associated with epilepsy, Parkinson's disease (Ma et al., 2021), and Alzheimer's disease due to its dysregulation in synaptic plasticity and neuroinflammatory processes (Ieguchi & Maru, 2019). EphA1's role in angiogenesis also makes it relevant to diseases such as diabetic retinopathy and age-related macular degeneration. Disruption of EphA1 signalling has been linked to age-related macular degeneration and neovascularization in diabetic retinopathy (Li et al., 2017). EphA1 regulates cytokine production, immune cell activation, and migration in immune-related disorders. Autoimmune diseases, including rheumatoid arthritis and inflammatory bowel disease, have also been linked to EphA1 dysregulation (Hjorthaug & Aasheim, 2007). Given its central role in tumour progression and angiogenesis, EphA1 represents an attractive therapeutic target. Inhibition of the ATP-binding pocket within the kinase domain offers a rational strategy to suppress EphA1-mediated oncogenic signalling. However, despite its biological importance, selective small-molecule inhibitors targeting EphA1 remain limited.
Structure-based drug design (SBDD) is a powerful strategy employed in drug development that uses the three-dimensional structure of a protein target to design small molecules with high affinity and specificity (Yu & MacKerell, 2017). With the potential to develop medicines with increased efficacy and fewer adverse effects, SBDD has become an essential approach in modern drug discovery. Virtual screening, a computational technique that efficiently identifies prospective lead compounds from large chemical libraries, is a key component of SBDD. By screening numerous compounds computationally, virtual screening allows the rapid identification of promising candidates for further experimental validation. This approach significantly streamlines the drug development process compared with traditional trial-and-error methods, thereby saving both time and cost.
In the present study, we employed an integrative computational strategy combining large-scale virtual screening, molecular docking, drug-likeness and ADMET profiling, PASS prediction, and molecular dynamics (MD) simulations to identify potential natural inhibitors of EphA1. A curated library of natural compounds from the ZINC database was screened against the EphA1 kinase domain to identify high-affinity binders targeting the ATP-binding site. The most promising candidates were further evaluated for pharmacokinetic suitability and dynamic stability to identify potential lead molecules for EphA1-targeted anticancer therapy.
2. Materials and Methods
2.1. Web Resources and the Computing Environment
The study was conducted on a Windows-based laptop equipped with a 1.19 GHz CPU, 8 GB RAM, and a 512 GB SSD. Throughout the research, a reliable power source and a high-speed internet connection were available. InstaDock (Mohammad et al., 2021) and Discovery Studio were utilized for virtual screening and molecular docking, while PyMOL (DeLano, 2002) and Discovery Studio Visualizer (Biovia, 2017) were used for visualization. Various resources and servers such as NCBI, UniProt (UniProt Consortium, 2019), the RCSB Protein Data Bank (Rose et al., 2017), AlphaFold (David et al., 2022), the ZINC database (Irwin & Shoichet, 2005; Irwin et al., 2020), SwissADME (Daina et al., 2017), pkCSM (Pires et al., 2015), Way2Drug PASS (Lagunin et al., 2000), and CarcinoPred-EL (Zhang et al., 2017) were utilized for data retrieval, evaluation, and analysis in this study. GROMACS was used to perform molecular dynamics simulations. The various systematic steps employed in this study are depicted in Figure.
2.2. Receptor and Library Preparation
The EphA1 structure was selected and retrieved from UniProt and AlphaFold based on structural completeness, the presence of the kinase domain, mutation-related sequence information, ligand-related information, and structural purity. The structure of EPHA1 was obtained in PDB format from the AlphaFold Protein Structure Database (David et al., 2022; UniProt Consortium, 2019) and further refined in PyMOL (DeLano, 2002) (Figure). Water molecules were removed during receptor preparation.
The ZINC database is a freely available public resource that compiles annotated, commercially available chemical structures (Irwin & Shoichet, 2005; Irwin et al., 2020). Both 2D and 3D structures of compounds can be downloaded from this database, and its interface supports rapid molecular lookup and analogue searching. ZINC maintains a catalogue of more than 230 million ready-to-dock 3D compounds for sale and also provides access to more than 750 million purchasable compounds, enabling rapid analogue searches (Irwin et al., 2020). The ZINC database has expanded substantially, and ZINC20 now contains 1.4 billion chemicals from 310 catalogues and 150 suppliers (Irwin et al., 2020). According to the 90/90/90 standard, 90% of catalogues are updated every 90 days, and 90% of compounds have been verified as available for purchase within the preceding three months (Irwin et al., 2020).
In the present study, the Lipinski rule of five was applied to select 32,901 small molecules from the ZINC database. The Lipinski rule is a cornerstone of medicinal chemistry and provides a set of threshold-based parameters to assess the drug-likeness of chemical compounds. Drug-like molecules generally satisfy the following criteria: molecular weight below 500 Da, fewer than 5 hydrogen-bond donors, fewer than 10 hydrogen-bond acceptors, and a LogP value below 5.
2.3. Molecular Docking and Virtual Screening
To investigate substrate inhibitor selectivity and understand the mechanism involved, high-throughput screening was conducted using the receptor-ligand docking method for the target AF-P21709-F1. The purpose was to examine the orientation of ligands within the protein's active site cavity. A collection of natural compounds was sourced from the ZINC database, and the ligands were acquired in pdbqt format. Molecular docking of EPHA1 was conducted to examine the bond conformations and binding affinity between the ligands and EPHA1. Regarding the docking process, a defined energy grid box with the dimensions of grid box centre (x = -1.511, y = 1.922, z = -35.295) and size parameters (X=56, Y=63, and Z=75) was used to ensure maximum binding affinity and obtain the best conformational pose for the protein-ligand interactions. The docking algorithm investigated different ligand orientations within the receptor's active site. Each ligand screening used a random seed generator and a scoring function to evaluate binding affinity.
After the virtual screening was completed, the docking results were analysed by examining the output files and log files. The most appropriate docked conformation was chosen for subsequent analysis. The PyMOL software (DeLano, 2002) and Discovery Studio Visualizer (Biovia, 2017) were employed to visualize and analyse the structure of the docked complex. Using the data as a starting point, we next went on to evaluate the compounds' potential as drugs utilising the Swiss-ADME server (http://www.swissadme.ch/) for PAINs analysis, Carcinogenicity using CarcinoPred-EL. Following this, we further evaluated the physicochemical properties, absorption, distribution, metabolism, and excretion of selected compounds using PCKCSM (http://biosig.unimelb.edu.au/pkcsm). Lastly, PASS analysis was performed using way2drug (http://way2drug.com/passonline/), where ligand activities with probabilities of activation greater than 0.71 were considered.
2.4. Drug-Likeness, PAINS, and ADMET Analysis
The shortlisted compounds were subjected to pan-assay interference compounds (PAINS) filtering using the SwissADME server to eliminate false positives. Additional physicochemical filters were applied based on total polar surface area (TPSA: 65–145 Å2) and solubility predictions. ADMET properties, including absorption, distribution, metabolism, excretion, and toxicity, were predicted using the pkCSM server. Gastrointestinal absorption, blood-brain barrier permeability, CYP2D6 inhibition, OCT2 substrate status, and AMES toxicity were evaluated. Carcinogenicity prediction was performed using CarcinoPred-EL.
2.5. PASS Analysis and Protein–Ligand Interaction Analysis
Prediction of Activity Spectra for Substances (PASS) analysis was performed using the Way2Drug PASS Online server to evaluate the potential biological activities of selected compounds. Activities with a probability of activity (Pa) greater than the probability of inactivity (Pi), particularly antineoplastic activity, were considered significant. Detailed interaction analysis of selected protein–ligand complexes was performed using Discovery Studio Visualizer. Hydrogen bonds, hydrophobic interactions, van der Waals interactions, and interactions with key catalytic residues within the ATP-binding pocket were examined. Special emphasis was placed on interactions with critical residues in the kinase domain, such as Lys656 and Asp749.
2.6. MD simulations
MD simulations were performed for 100 nanoseconds on both free EphA1 and EphA1 with the compound ZINC12660859 using GROMACS 5.1.2 at 300 Kelvin. The simulations utilized the GROMACS G54a7 force field at the molecular mechanics level. The systems were immersed in a cubic box of water (10 Å) and solvated with the spc216 water model using the gmx solvate module. Energy minimization was performed using 1500 steepest-descent steps. During the equilibration phase, the temperature gradually increased from 0 to 300 Kelvin over 100 picoseconds, while maintaining constant volume and periodic boundary conditions. Trajectory analysis employed various GROMACS utilities, including gmx rmsd for root-mean-square deviation, gmx rmsf for root-mean-square fluctuation, gmx gyrate for radius of gyration, gmx sasa for solvent-accessible surface area, and gmx hydrogen-bonds for intra- and intermolecular hydrogen bonds. Graphs and figures were generated using QtGrace.
3. Results and Discussion
3.1. Virtual Screening and Binding Affinity Analysis
We conducted virtual screening of compounds to identify a competitive inhibitor for EphA1. Log files and out-files were created, containing affinity scores and docked poses for each compound in the directory. Based on binding affinities, docking scores, and binding poses, we filtered compounds using the log and output files. Our screening identified several natural compounds with favourable binding affinity scores for EphA1's binding pocket, indicating their potential as EphA1 inhibitors. Out of 32,901 compounds that were screened from the output, 320 were found to have significant EphA1 binding affinity scores ranging from -11.2 to -9.4 (Table). Among the screened compounds, ZINC03845566 exhibited the highest binding affinity (-11.2 kcal/mol), while ZINC12660859 and ZINC12661003 demonstrated binding energies of -9.9 and -9.7 kcal/mol, respectively. Although several compounds showed slightly stronger docking scores, subsequent pharmacokinetic filtering and biological activity prediction narrowed the focus to ZINC12660859 and ZINC12661003 due to their favorable drug-like properties and predicted antineoplastic potential. The observed binding energies suggest strong stabilization within the EphA1 catalytic pocket, indicating potential competitive inhibition at the ATP-binding site.
3.2. Drug Likeness and ADMET Analysis
In the initial step, Discovery Studio was used to generate SMILES strings for the top hits. These SMILES IDs were subsequently subjected to PAINS analysis using SwissADME. The PAINS analysis aimed to identify pan-assay interference compounds, which are multi-target ligands that lack specificity for a single target. Eliminating such compounds is crucial because they can lead to unintended regulatory effects. Following the PAINS analysis, additional filters were applied to refine the compound selection process. Compounds violating Lipinski’s rule of five were removed. Furthermore, compounds with a total polar surface area (TPSA) outside the range of 65 to 145 were excluded. Compounds exhibiting poor solubility according to Silicos-IT Solubility (mol/l), Ali Solubility (mol/l), and ESOL Solubility (mol/l) were also eliminated from consideration. These filtering criteria helped in identifying the most promising compounds for further analysis and evaluation (Table).
After applying the filters, 24 compounds remained for further analysis. Using pkCSM, these substances were screened to determine their ADMET parameters. Remarkably, 20 compounds successfully met the ADMET parameters, indicating favourable characteristics for drug development. The physicochemical parameters evaluated for these compounds were found to fall within the range deemed suitable for drug candidacy, as outlined in Table. This observation indicates that the selected compounds possess desirable properties, making them promising candidates for further investigation and drug development.
In addition, a PASS analysis was performed on the 20 selected compounds using SMILES strings via the way-2-drugs-PASS Online tool. This analysis aimed to identify compounds with anti-cancer activity, thereby offering therapeutic leads for EphA1 targeting. After careful analysis, two compounds were chosen based on their demonstrated anti-cancer properties. The activities and corresponding Pa (probability of activity) and Pi (probability of inactivity) values for these selected compounds are presented in Table. These compounds exhibit promising characteristics and warrant further exploration as potential anti-cancer agents targeting EphA1.
3.3. Interaction Analysis
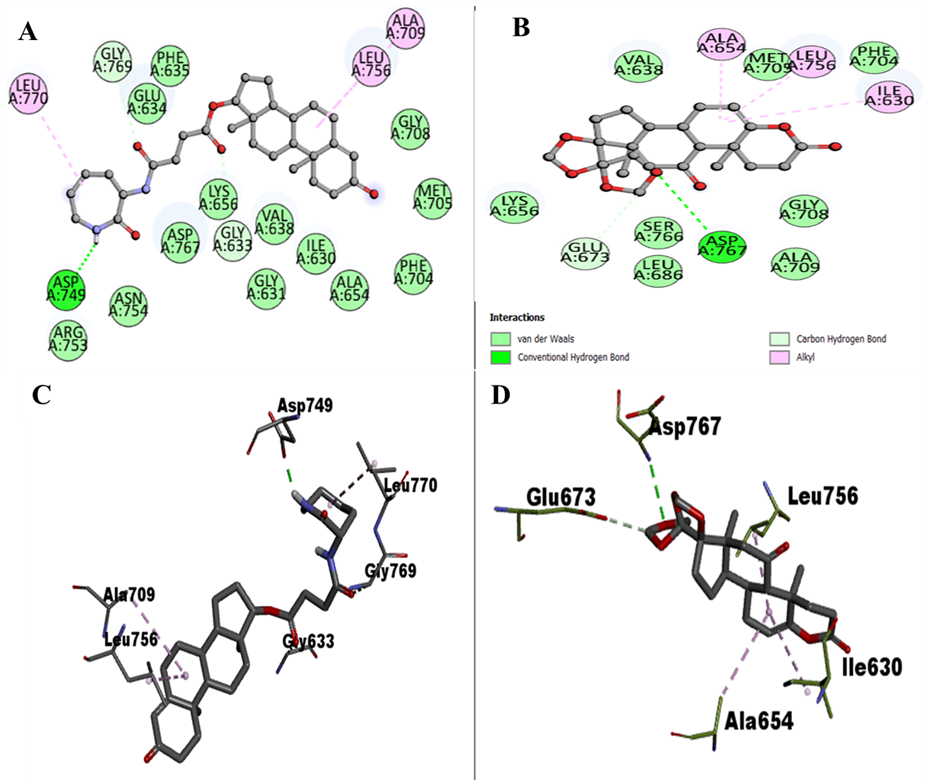
To analyse the binding modes and interaction patterns of the two selected compounds, Discovery Studio was used. A set of 18 docked conformers was produced for each of the two natural compounds using the output files. The analysis focused on examining the interacting residues within the compounds. The Discovery Studio software was used to identify and visualize hydrogen bonding and other interactions between the compounds and EphA1. Notably, residues within the kinase domain of EphA1 were observed to play a significant role in mediating numerous interactions with the compounds. This analysis provides valuable insights into the binding mechanism and potential interactions between the selected compounds and EphA1. Residues of EphA1's kinase domain have been shown to offer a considerable number of interactions, including Asp749, Gly769, Gly633, Asp767, Glu673, Lys656, Arg753, Asn754, Val638, Ile630, Leu,770, Leu756, Ala709, Gly631, Ala654, Phe704, Met705, Gly708, Phe635, Glu634, Ser766, Leu686, Gly708 (Table). Each compound interacts with ATP binding site or is closely associated with ATP binding sites. ATP binding sites, Lys656, as of EphA1, are inhibited either by a hydrogen bond or a strong bond in each compound, and active site Asp749 of EphA1 closely forms bonds with compounds.
Compound ZINC12660859 is forming hydrogen bonds to Asp749, Gly769, Gly633 and several other interactions to656, Arg753, Asn754, Asp767, Val638, Ile630, Leu,770, Leu756, Ala709, Gly631, Ala654, Phe704, Met705, Gly708, Phe635, Glu634. Compound ZINC12661003 is interacting to GlAsp767, Glu673 via Hydrogen bonding, and Lys656, Ser766, Leu686, Ala709, Gly708, Val638, Met705, Phe704, Ala654, Leu756, Ile630 are participating in other interactions (Figure). A significant number of interactions, including Hydrogen bonding with the active site Asp 749 and Vander wall’s interaction with ATP binding site Lys656, formed between EphA1 and ZINC12660859, demonstrating a significant affinity for binding and its further implications as a medicinal compound targeting EphA1 (Figure).
3.4. MD Simulations
An effective way to examine the atomic-level structural specifics and dynamic behaviour of protein-ligand complexes is to use the MD simulation approach. To acquire a thorough understanding of the stability and dynamics of the EphA1-ZINC12660859 complex, we conducted all-atom MD simulations for a duration of 100 ns on both the EphA1-ZINC12660859 complex and the apo EphA1 protein. We aimed to uncover numerous systematic and structural factors that affect the complex's behaviour through rigorous research into intermolecular interactions, conformational changes, hydrogen-bonding patterns, and solvent effects. Our research contributes to the field of drug development and targeted therapies by providing a deeper understanding of atomic-level protein-ligand interactions.
Root-Mean-Square Deviation
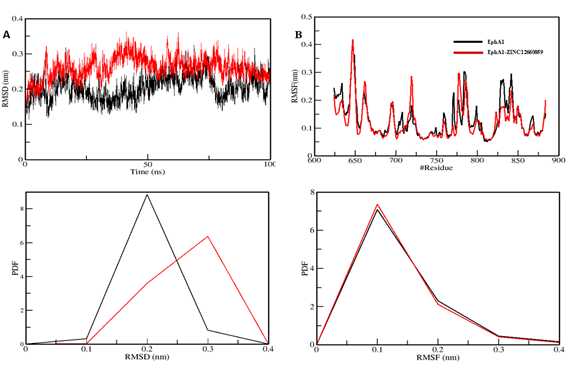
The root-mean-square deviation (RMSD) is a well-known metric frequently used to analyse structural heterogeneity in proteins. It is crucial as a tool for assessing and analysing the dynamic behaviour and conformational changes of protein structures. RMSD provides vital insights into protein structural deviation and dynamics by quantifying the discrepancy between the atomic positions of a target structure and those of a reference structure. The structural dynamics of the protein EphA1 before and after ligand binding revealed low mean RMSD values of 0.20 nm and 0.26 nm for EphA1 and EphA1-ZINC12660859, respectively (Figure A). During the 100 ns simulation, both systems exhibited stable trajectories, with RMSD values approaching equilibrium. Analysis of the probability distribution function (PDF) revealed a significant increase in EphA1 stability, as evidenced by a pronounced peak in the distribution during compound binding. This finding indicates that the binding of ZINC12660859 positively influenced the overall stability of the complex.
Root-Mean Square Fluctuation
The root mean square fluctuation (RMSF) analysis has been widely employed to investigate protein dynamics during MD simulations. It offers valuable information on the influence of ligand binding on protein vibrations. Through the examination of the RMSF plot, we examined the residual dynamics of EphA1 prior to and following ligand binding. Notably, the protein-ligand complex exhibited a significant decrease in RMSF fluctuations, indicating increased stability upon binding of ZINC12660859 (Figure B). The RMSF analysis revealed a significant decrease in residual fluctuation within the POT1 protein. These findings are further corroborated by PDF analysis, which clearly indicates a pronounced decrease in residual flexibility following complex formation.
Radius of gyration
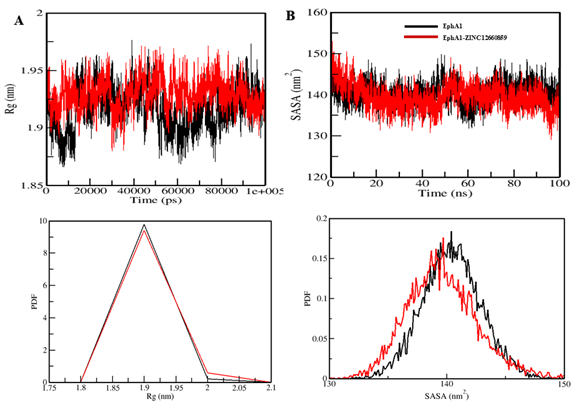
The radius of gyration (Rg) is a direct indicator of a protein's tertiary structure and overall conformation. It is commonly used to assess the compactness and folding behaviour of proteins. In our study, we investigated the stability of both EphA1 and the EphA1-ZINC12660859 complex by analysing their respective Rg values. The average Rg for EphA1 in its apo form was 1.92 nm, whereas for the EphA1-ZINC12660859 complex, it was found to be 1.93 nm (Figure A). The Rg plot revealed a slight increase of approximately 0.01 nm in Rg upon binding of ZINC12660859 to EphA1. This minor increase is likely due to the compound's packing effect. Remarkably, the introduction of ZINC12660859 did not result in any notable alterations to the structure of EphA1. Throughout the simulation trajectory, EphA1 maintained a constant equilibrium of Rg, indicating the enduring stability of the complex.
Solvent Accessible Surface Area
The solvent-accessible surface area (SASA) of a protein refers to the region on its surface that is accessible to the surrounding solvent. Analysing SASA is an important method for examining protein folding and stability. Upon analysis of the SASA plot, no notable alterations in SASA values were observed, indicating the stability of EphA1 interactions with ZINC12660859 (Figure B). The SASA values for the EphA1 and EphA1-ZINC12660859 complexes were 140.42 nm2 and 139.62 nm2, respectively. The distribution of surface-area values exhibited a consistent pattern across both systems. Notably, there was a marginal reduction in the average surface area upon binding of the compound to EphA1. This reduction suggests the formation of stable interactions or binding between the protein and ligand.
Hydrogen Bond Analysis
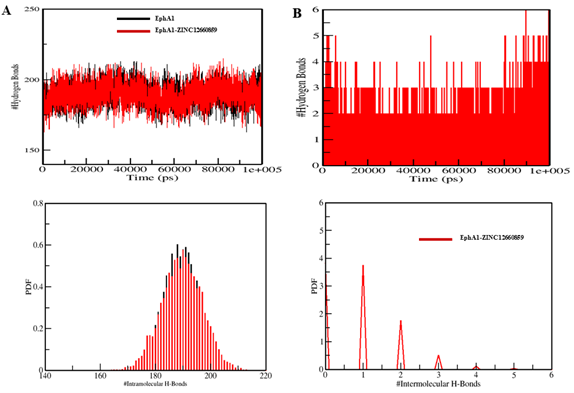
The creation of hydrogen bonds (H-bonds) is critical in the kinetics of protein folding. Protein conformational alterations are primarily governed by the disruption and formation of hydrogen bonds. In this study, we analysed the temporal progression of hydrogen bonds to assess the integrity of intramolecular bonding within the EphA1-ZINC12660859 complex. The plot we obtained indicated negligible alterations in the number of hydrogen bonds in EphA1 upon complex formation with ZINC12660859 (Figure). In the context of EphA1 intramolecular interactions, we observed that the average number of hydrogen bonds formed was 189 before binding of ZINC12660859, and this count increased to 190 after binding. A minor rise in the number of hydrogen bonds was noted, which can be attributed to the heightened compactness resulting from the binding of the ligand (Figure A). Analysis of the PDFs for both systems confirmed the consistent stability of intramolecular hydrogen bonds. From the generated plots, it can be inferred that the intramolecular hydrogen bonds in EphA1 remained stable throughout the simulation, even after compound binding. Throughout the simulation, ZINC12660859 was discovered to bind into the specified pocket of EphA1, creating 1–2 stable hydrogen bonds with little variation (Figure B). There were further occasions where ZINC12660859 showed 3–4 hydrogen bonds with larger variations. ZINC12660859 remained at its original docking site on EphA1, consistent with our predictions based on the observed intermolecular hydrogen bonding. These hydrogen bonds play an important role in the stability of the complex structure, preventing the compound from migrating or dissociating from its binding site on EphA1 during the simulation.
4. Conclusions
In the present study, an integrative structure-based computational approach was employed to identify potential natural inhibitors of the EphA1 receptor tyrosine kinase. Large-scale virtual screening of 32,901 drug-like natural compounds led to the identification of promising candidates targeting the ATP-binding pocket of the EphA1 kinase domain. Among the shortlisted molecules, ZINC12660859 demonstrated favorable binding affinity, compliance with drug-likeness criteria, acceptable ADMET properties, predicted antineoplastic activity, and stable interactions with key catalytic residues, including Lys656 and Asp749. Molecular dynamics simulations further confirmed the structural stability and sustained binding of the EphA1–ZINC12660859 complex over 100 ns without inducing conformational destabilization. Collectively, these findings suggest that ZINC12660859 represents a promising lead compound for EphA1-targeted inhibition. While the present work provides strong computational evidence supporting its therapeutic potential, further in vitro and in vivo experimental validation is required to confirm its inhibitory activity and anticancer efficacy. This study highlights the effectiveness of structure-based virtual screening and molecular dynamics simulations in accelerating the discovery of kinase-targeted therapeutic candidates.
Conflict of Interest
There is no conflict of interest to declare.
Data Availability Statement
The data supporting this study are provided in this article.
Funding
None
Acknowledgements
None
Supplementary Materials
None
Figures
Figure 1. Steps involved in methodology
{kind=link}
Figure 2. Kinase domain of EphA1. The structure was prepared in PyMOL.
{kind=link}
Figure 3. 2D interactions of EphA1 with (A) ZINC12660859, (B) ZINC12661003, (C) representation of amino acid residues of EphA1 interacting with ZINC12660859, and (D) ZINC12661003.
{kind=link}
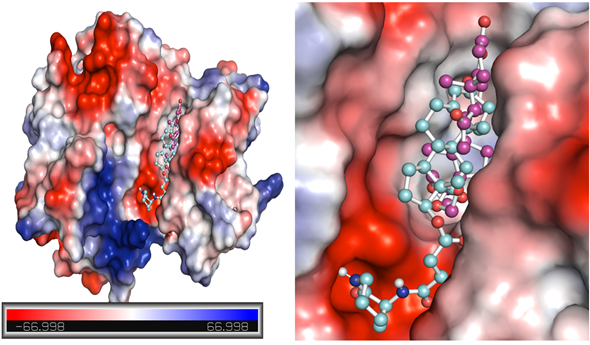
Figure 4. Potential surface representation of EphA1 complex with ZINC12660859 (cyan) and ZINC12661003 (magenta).
{kind=link}
Figure 5. Structural dynamics of EphA1-ZINC12660859 complex. (A) RMSD plot. (B) RMSF plot. The lower panels show the probability distribution functions (PDFs).
{kind=link}
Figure 6. Analysis of the structural compactness and folding of EphA1 upon interaction with ZINC12660859. The changes in radius of gyration (Rg) and solvent-accessible surface area (SASA) are presented in plots A and B, respectively.
{kind=link}
Figure 7. Analysis of hydrogen bonds. (A) Intramolecular H-bonds' temporal progression. (B) Intermolecular H-bonds established between EphA1 and ZINC12660859 within 0.35 nm. The PDF values for the hydrogen-bond distribution are shown in the lower panels. # stands for number.
{kind=link}
References
- 1
Adu-Gyamfi, E.A., Czika, A., Liu, T.H., et al. 2021, Biology of Reproduction, 104, 71–82, doi: 10.1093/biolre/ioaa171 DOI
- 2
Biovia, D.S. 2017, Discovery Studio Visualizer
- 3
Dai, B.Q., & Zhang, X. 2020, Neoplasma, 67, 794–801, doi: 10.4149/neo_2020_190807N724 DOI
- 4
Daina, A., Michielin, O., Zoete, V. 2017, Scientific Reports, 7, 42717, doi: 10.1038/srep42717 DOI
- 5
David, A., Islam, S., Tankhilevich, E., Sternberg, M.J.E. 2022, Journal of Molecular Biology, 434, 167336, doi: 10.1016/j.jmb.2021.167336 DOI
- 6
DeLano, W.L. 2002, CCP4 Newsletter on Protein Crystallography, 40, 82–92
- 7
Gajdzis, M., Theocharis, S., Gajdzis, P., et al. 2020, Life, 10, doi: 10.3390/life10100225 DOI
- 8
Hjorthaug, H.S., & Aasheim, H.C. 2007, European Journal of Immunology, 37, 2326–2336, doi: 10.1002/eji.200737111 DOI
- 9
Ieguchi, K., & Maru, Y. 2019, Cancer Science, 110, 841–848, doi: 10.1111/cas.13942 DOI
- 10
Irwin, J.J., & Shoichet, B.K. 2005, Journal of Chemical Information and Modeling, 45, 177–182, doi: 10.1021/ci049714+ DOI
- 11
Irwin, J.J., Tang, K.G., Young, J., et al. 2020, Journal of Chemical Information and Modeling, 60, 6065–6073, doi: 10.1021/acs.jcim.0c00675 DOI
- 12
Kang, M., Jeong, W., Bae, H., et al. 2018, Journal of Cellular Physiology, 233, 2560–2571, doi: 10.1002/jcp.26131 DOI
- 13
Lagunin, A., Stepanchikova, A., Filimonov, D., Poroikov, V. 2000, Bioinformatics, 16, 747–748, doi: 10.1093/bioinformatics/16.8.747 DOI
- 14
Li, Y., Yan, H., Wang, F., et al. 2017, Biochemical and Biophysical Research Communications, 486, 693–699, doi: 10.1016/j.bbrc.2017.03.100 DOI
- 15
Liang, Z., Wang, X., Dong, K., et al. 2021, BioMed Research International, 2021, 5575704, doi: 10.1155/2021/5575704 DOI
- 16
Lisabeth, E.M., Falivelli, G., Pasquale, E.B. 2013, Cold Spring Harbor Perspectives in Biology, 5, doi: 10.1101/cshperspect.a009159 DOI
- 17
Ma, J., Wang, Z., Chen, S., et al. 2021, Molecular Neurobiology, 58, 913–925, doi: 10.1007/s12035-020-02122-x DOI
- 18
Mohammad, T., Mathur, Y., Hassan, M.I. 2021, Briefings in Bioinformatics, 22, doi: 10.1093/bib/bbaa279 DOI
- 19
Peng, L., Wang, H., Dong, Y., et al. 2013, International Journal of Clinical and Experimental Pathology, 6, 1854–1860
- 20
Pires, D.E., Blundell, T.L., Ascher, D.B. 2015, Journal of Medicinal Chemistry, 58, 4066–4072, doi: 10.1021/acs.jmedchem.5b00104 DOI
- 21
Rose, P.W., Prlic, A., Altunkaya, A., et al. 2017, Nucleic Acids Research, 45, D271–D281, doi: 10.1093/nar/gkw1000 DOI
- 22
Toma, M.I., Erdmann, K., Diezel, M., et al. 2014, PLoS One, 9, e102262, doi: 10.1371/journal.pone.0102262 DOI
- 23
UniProt Consortium 2019, Nucleic Acids Research, 47, D506–D515, doi: 10.1093/nar/gky1049 DOI
- 24
Villegas-Llerena, C., Phillips, A., Garcia-Reitboeck, P., et al. 2016, Current Opinion in Neurobiology, 36, 74–81, doi: 10.1016/j.conb.2015.10.004 DOI
- 25
Wang, J., Ma, J., Dong, Y., et al. 2013, APMIS, 121, 30–37, doi: 10.1111/j.1600-0463.2012.02941.x DOI
- 26
Wang, Y., Yu, H., Shan, Y., et al. 2016, Journal of Experimental & Clinical Cancer Research, 35, 65, doi: 10.1186/s13046-016-0339-6 DOI
- 27
Wang, Y.C., Dai, Y., Xu, G.L., et al. 2020, Medical Science Monitor, 26, e923409, doi: 10.12659/msm.923409 DOI
- 28
Wu, B.O., Jiang, W.G., Zhou, D., Cui, Y.X. 2016, Anticancer Research, 36, 1211–1219
- 29
Wu, Y., Du, Z., Mou, J., et al. 2022, Current Medicinal Chemistry, doi: 10.2174/0929867329666220820125638 DOI
- 30
Yamazaki, T., Masuda, J., Omori, T., et al. 2009, Journal of Cell Science, 122, 243–255, doi: 10.1242/jcs.036467 DOI
- 31
Yu, L., Ke, J., Du, X., et al. 2019, Scientific Reports, 9, 2369, doi: 10.1038/s41598-019-38878-z DOI
- 32
Yu, W., & MacKerell, A.D., Jr. 2017, Methods in Molecular Biology, 1520, 85–106, doi: 10.1007/978-1-4939-6634-9_5 DOI
- 33
Zhang, L., Ai, H., Chen, W., et al. 2017, Scientific Reports, 7, 2118, doi: 10.1038/s41598-017-02365-0 DOI
Tables
Table 1. List of selected top 66 compounds based on binding affinity towards EphA1.
| |c|l|c| S. No. | Ligand ZINC ID | Binding Free Energy (kcal/mol) | S. No. | Ligand ZINC ID | Binding Free Energy (kcal/mol) |
|---|---|---|---|---|---|
| 1 | ZINC03845566 | -11.2 | 34 | ZINC04017374 | -10.5 |
| 2 | ZINC08790757 | -11.1 | 35 | ZINC08789996 | -10.5 |
| 3 | ZINC08791024 | -11.1 | 36 | ZINC08876667 | -10.5 |
| 4 | ZINC08791220 | -11.1 | 37 | ZINC12873131 | -10.5 |
| 5 | ZINC08791221 | -11.1 | 38 | ZINC12874710 | -10.5 |
| 6 | ZINC12878012 | -11.0 | 39 | ZINC12886094 | -10.5 |
| 7 | ZINC12902062 | -11.0 | 40 | ZINC12887931 | -10.5 |
| 8 | ZINC12877669 | -10.9 | 41 | ZINC12902067 | -10.5 |
| 9 | ZINC12902057 | -10.9 | 42 | ZINC08918297 | -10.4 |
| 10 | ZINC12876960 | -10.8 | 43 | ZINC02161110 | -10.4 |
| 11 | ZINC12881190 | -10.8 | 44 | ZINC04258868 | -10.4 |
| 12 | ZINC08789288 | -10.8 | 45 | ZINC08300419 | -10.4 |
| 13 | ZINC08790429 | -10.8 | 46 | ZINC08789048 | -10.4 |
| 14 | ZINC08876663 | -10.8 | 47 | ZINC12885214 | -10.4 |
| 15 | ZINC12883374 | -10.8 | 48 | ZINC03841970 | -10.4 |
| 16 | ZINC08918144 | -10.8 | 49 | ZINC08789855 | -10.4 |
| 17 | ZINC08791168 | -10.8 | 50 | ZINC12887928 | -10.4 |
| 18 | ZINC08790034 | -10.7 | 51 | ZINC08792436 | -10.4 |
| 19 | ZINC08789997 | -10.7 | 52 | ZINC08790023 | -10.4 |
| 20 | ZINC08790428 | -10.7 | 53 | ZINC03851871 | -10.3 |
| 21 | ZINC09033817 | -10.6 | 54 | ZINC08918422 | -10.3 |
| 22 | ZINC08790759 | -10.6 | 55 | ZINC08918466 | -10.3 |
| 23 | ZINC08790760 | -10.6 | 56 | ZINC12662629 | -10.3 |
| 24 | ZINC08876661 | -10.6 | 57 | ZINC03844856 | -10.3 |
| 25 | ZINC12879301 | -10.6 | 58 | ZINC12895934 | -10.3 |
| 26 | ZINC02133362 | -10.6 | 59 | ZINC04045796 | -10.2 |
| 27 | ZINC08876556 | -10.6 | 60 | ZINC04073739 | -10.2 |
| 28 | ZINC11851754 | -10.6 | 61 | ZINC12874870 | -10.2 |
| 29 | ZINC12662242 | -10.6 | 62 | ZINC00348369 | -10.2 |
| 30 | ZINC12902051 | -10.6 | 63 | ZINC02090582 | -10.2 |
| 31 | ZINC08791169 | -10.6 | 64 | ZINC04237100 | -10.2 |
| 32 | ZINC03850412 | -10.5 | 65 | ZINC12660859 | -9.9 |
| 33 | ZINC08876707 | -10.5 | 66 | ZINC12661003 | -9.7 |
Table 2. The predicted druglike properties of four potentially selective inhibitors of EphA1.
| Compound ID | Molecular Weight | Rotatable | ||||
|---|---|---|---|---|---|---|
| Bonds | H-Bond | |||||
| Donor | H-Bond | |||||
| Acceptor | LogP | PAINS | ||||
| ZINC12660859 | 498.664 | 5 | 2 | 5 | 3.9952 | 0 |
| ZINC12661003 | 404.459 | 0 | 0 | 7 | 2.6825 | 0 |
Table 3. Predicted ADMET characteristics of chosen hits.
| ZINC ID | Absorption | |||
|---|---|---|---|---|
| GI | ||||
| Absorption | Distribution | |||
| BBB | ||||
| Permeation | Metabolism | |||
| CYP2D6 | ||||
| Inhibitor | Excretion | |||
| OCT2 | ||||
| Substrate | Toxicity | |||
| AMES | ||||
| ZINC12660859 | 94.952 | |||
| (High) | -0.177 | No | No | No |
| ZINC12661003 | 99.476 | |||
| (High) | -0.653 | No | No | No |
Table 4. PASS analysis results: Activities with Pa and Pi values of 2 selected compounds.
| S. No. | Compound | Pa value | Pi value | Biological Activity |
|---|---|---|---|---|
| μltirow3*1. | μltirow3*ZINC12660859 | 0,845 | 0,025 | CYP2C12 substrate |
| 3-5 | 0,500 | 0,027 | AR expression inhibitor | |
| 3-5 | 0,464 | 0,083 | Antineoplastic | |
| μltirow5*2. | μltirow5*ZINC12661003 | 0,566 | 0,053 | Antineoplastic |
| 3-5 | 0,649 | 0,027 | Phosphatase inhibitor | |
| 3-5 | 0,488 | 0,016 | Myc inhibitor | |
| 3-5 | 0,388 | 0,079 | Apoptosis agonist | |
| 3-5 | 0,339 | 0,066 | Antimetastatic |
Table 5. Interacting residues analyzed using Discovery Studio.
| |p7cm| S. No. | Compound ID | Interacting Residues | |
|---|---|---|---|
| 3-4 | H BONDING | Other Interactions | |
| 1. | ZINC12660859 | Asp749, Gly769, Gly633 | Lys656, Arg753, Asn754, Asp767, Val638, Ile630, Leu770, Leu756, Ala709, Gly631, Ala654, Phe704, Met705, Gly708, Phe635, Glu634 |
| 2. | ZINC12661003 | Asp767, Glu673 | Lys656, Ser766, Leu686, Ala709, Gly708, Val638, Met705, Phe704, Ala654, Leu756, Ile630 |
Table 6. The mean values of various MD parameters were computed based on 100 ns simulations.
| System | RMSD | ||||
|---|---|---|---|---|---|
| (nm) | RMSF | ||||
| (nm) | Rg | ||||
| (nm) | SASA | ||||
| (nm2) | #H- | ||||
| Bonds | |||||
| EphA1 | 0.20 | 0.13 | 1.92 | 140.42 | 189 |
| EphA1-ZINC12660859 | 0.26 | 0.12 | 1.93 | 139.62 | 190 |