


About Article
Elucidating impacts of deleterious Missense mutations on the structure and function of β-glucuronidase: Investigating the molecular basis of pathogenesis of MPSVII
Volume 1, No. 1 · 2026
Highlights
- A comprehensive in silico study was carried out on missense variants in the GUSB gene, which encodes the lysosomal enzyme β-glucuronidase implicated in MPS VII (Sly syndrome).
- The article reports integrative sequence-based screening with SIFT, PolyPhen-2, Mutation Assessor, and FATHMM, combined with structure-based assessment using mCSM, MAESTROweb, PremPS, and DynaMut.
- Fifteen mutations were prioritized as deleterious and structurally destabilizing, and eight variants were further highlighted as high-confidence pathogenic mutations.
- SODA-based solubility analysis suggested that three of the eight prioritized pathogenic variants are less soluble and more aggregation-prone.
- Structural comparison particularly emphasized mutations at residues 566 and 388, where loss of hydrogen bonding, aromatic contacts, and hydrophobic interactions was linked to destabilization.
Abstract
The rapid expansion of genome sequencing projects has resulted in the identification of numerous hypothetical proteins whose functions remain uncharacterized. In Listeria monocytogenes serotype 4b, a major food-borne pathogen associated with high mortality rates, several predicted proteins lack functional annotation despite their potential role in pathogenicity and survival. In the present study, a comprehensive in silico approach was employed to functionally annotate 92 hypothetical proteins identified from the genome of L. monocytogenes. Protein sequences were retrieved and analyzed using sequence similarity searches, conserved domain identification, motif analysis, and multiple sequence alignment. Functional classification was performed based on BLAST, Pfam, and InterPro analyses. Structural prediction was performed via homology modeling via the SWISS-MODEL server, followed by structural validation and comparative analysis using PyMOL and DALI-Lite. Functional inference from structural models was supported by conserved-residue mapping and ProFunc analysis. Sequence-based analysis enabled classification of most hypothetical proteins into functional groups, including hydrolases, transferases, transporters, kinases, stress-response proteins, membrane proteins, DNA-binding proteins, and ATP-binding proteins. Structure-based modeling of selected proteins further confirmed the predicted catalytic residues, metal-binding sites, ligand-interaction sites, and conserved functional motifs. Several proteins were predicted to be involved in enzymatic activity, nucleotide metabolism, membrane transport, transcriptional regulation, and stress adaptation. This integrative computational analysis provides functional insights into previously uncharacterized proteins of L. monocytogenes. The findings enhance genome annotation quality and identify potential targets for further experimental validation, contributing to a better understanding of bacterial physiology and pathogenesis.
Keywords
Article Overview
This article examines how deleterious missense variants in the GUSB gene may alter β-glucuronidase stability and function, thereby contributing to mucopolysaccharidosis type VII (MPS VII).
The introduction contextualizes GUSB within lysosomal glycosaminoglycan degradation and links its dysfunction to lysosomal storage pathology, extracellular-matrix remodeling, and broader disease mechanisms.
The paper positions computational variant prioritization as a practical strategy for distinguishing pathogenic nsSNPs from benign variation and for relating mutation data to structural mechanisms.
1. About the Article
This is a research article published in Clinical & Molecular Biomedicine that focuses on the molecular consequences of missense mutations in the GUSB gene.
The work is framed around MPS VII, also called Sly syndrome, and combines variant collection, pathogenicity prediction, structural stability assessment, conservation analysis, and aggregation-propensity analysis.
- Journal: Clinical & Molecular Biomedicine
- Volume/Issue: Vol. 1, No. 1
- Header year: 2026
- Authors: Nushrat Jahan; Barka Basharat; Shakilur Rahman
- Affiliation 1: Department of Biotechnology, School of Chemical and Life Sciences, Jamia Hamdard, New Delhi, India
- Affiliation 2: Biology Department, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia
- Corresponding author: Dr. Shakilur Rahman
- Corresponding email: srahman@imamu.edu.sa
- Received: April 16th, 2024
- Accepted: March 31, 2026
- Published: May 21, 2024
- License: Creative Commons CC-BY 4.0
- Open-access status: Open Access
- Header page range on first page: pp. 1–25
- Printed article page range visible in PDF pages: 1–8
2. Introduction
1. Introduction1 𝛽-glucuronidase (GUSB) is a crucial enzyme widely used in 2 molecular biology and genetic engineering projects. (Yang et al.,3 2017). 3 The housekeeping enzyme GUSB plays an active role in 4 proteoglycan synthesis in lysosomes and is expressed in most5 tissues. It contributes significantly to the gradual decline in 6 dermatan and keratan sulfates by enhancing the disintegration7 of GAGs during the fifth cycle. GUSB promotes the dispersion of8 active or inert compounds from glucuronides, thereby modifying9 the manner in which prodrugs respond and operate (Naz et al.,10 2013).11 GUSB deficiency leads to mucopolysaccharidosis type VII,12 resulting in brain lysosomal storage (Kong et al., 2022). Humans13 have been shown to exhibit 11 distinct types (MPS I to IIIA, B,14 C, and IV to IX), which are divided according to the deficient15 enzyme (Hytönen et al., 2012). There are seven different types16 of Mucopolysaccharidosis (MPS), which are categorized based17 on defects in a certain number of the eight specific lysosomal18 metabolic enzymes (Khan et al., 2017). Mutations in the IDUA19 gene on chromosome 4p16 cause the first kind of MPS, which 20 is caused by a deficiency of the enzyme a-l-iduronidase. A-l- 21 iduronidase deficiency results in the accumulation of GAGs, such 22 as dermatan and heparan sulphates (DS and HS), in many human 23 tissues, leading to severe organ malfunction (Nagpal et al., 2022). 24 MPS II is linked to lysosomal dysfunction due to a deficiency 25 of the enzyme iduronate 2-sulphatase, which degrades heparan 26 sulphate and dermatan sulphate (DS). A rapidly developing 27 neurodegenerative lysosomal storage disease is MPSIII. It is 28 caused by biallelic variants in a gene encoding an enzyme in 29 the digestive tract that degrades heparan sulfate. Neuronal 30 inflammation and significant activation of astrocytes and 31 microglia are hallmarks of MPS III, a neurological disease 32 (Seker Yilmaz et al., 2021). MPS IVA, additionally referred 33 to as Morquio syndrome type A, constitutes one of the most 34 significant lysosomal illnesses. The lysosomal hydrolase N- 35 acetylglucosamine-6-sulfate sulfatase (GALNS) is absent in this 36 autosomal-recessive genetic disorder (Sawamoto et al., 2020). 37 There is nothing abnormal regarding the nervous system’s 38 functions, but common features include elevated blood and urine 39 Copyright: Distributed under Creative Commons CC-BY 4.0 OPEN ACCESS 1–8 Elucidating impacts of deleterious Missense mutations on the structure and function of 𝛽-glucuronidase: Investigating the molecular basis of pathogenesis of MPSVII potassium (K+) levels, a petite stature, odontoid hypoplasia,40 pectus carinatum, kyphoscoliosis, genu valgum, joint laxity, and41 corneal clouding (Tomatsu et al., 2011).42 Many lysosomal storage disorders, including43 mucopolysaccharidosis type VII (MPS VII), commonly referred44 to as Sly syndrome, are frequently linked to these mutations (?).45 The integrity and activity of 𝛽-glucuronidase are dramatically46 affected by missense, frameshift, and nonsense mutations in the47 GusB gene, according to recent investigations (?). GusB Gene48 encodes a 651-amino-acid-long homotetrameric protein with49 three distinctive domains in every monomer (Florindo et al.,50 2018). One such relationship involves the lysosomal-associated51 membrane protein 2 (LAMP2), which supports the stability and52 appropriate transit of 𝛽-glucuronidase inside lysosomes (Staudt53 et al., 2016). Furthermore, heparan sulfate and chondroitin54 sulfate are broken down by 𝛽-glucuronidase, which works55 in tandem with other enzymes such as arylsulfatase B and56 heparanase (Naz et al., 2013).57 According to Urayama et al. (2004), it is imperative that58 GUSB reach its intended intracellular location via this targeting59 mechanism. The lysosomal route of GAG breakdown is the60 main pathway in which GUSB is involved. An essential stage 61 in the breakdown of GAGs is the cleavage of the 𝛽-D-glucuronic62 acid residues by GUSB. GAG buildup is caused by a deficit in 𝛽-63 glucuronidase activity and results in lysosomal storage disorders64 (Hytönen et al., 2012). GAGs (glycosaminoglycans) are broken65 down by many lysosomal enzymes in an intricate procedure. The66 remaining molecules of 𝛽-D-glucuronic acid from GAGs such as67 heparan sulfate, dermatan sulfate, and chondroitin sulfate are68 particularly susceptible to hydrolysis by GUSB (Awolade et al.,69 2020).70 GUSB utilizes its N-terminus domain to identify and adhere 71 to a GAG chain, its substrate. The catalytic function of the 72 enzyme in question depends on the initial binding (Urayama73 et al., 2004). The site that catalyzes the breakdown of 𝛽-D-74 glucuronic acid residues is located in the catalytic region of GUSB75 (Hytönen et al., 2012). After splitting, the active region of the76 enzyme discharges the resulting molecules, which are shorter77 oligosaccharides, enabling 𝛽-Glucuronidase to interact with a 78 different substrate (Staudt et al., 2016). Supporting autophagy 79 and lysosomal activity requires 𝛽-glucuronidase. It supports the80 prevention of cellular malfunction by facilitating the breakdown81 of autophagic organelles. The amino acids associated with LAMP282 maintain its continued existence within lysosomes and ensure a83 steady supply of functional GUSB for the degradation of lysosomal 84 proteins (Staudt et al., 2016).85 GUSB is involved in the processes of extracellular matrix86 reorganization and cell-mediated cascade modulation, in addition87 to lysosomal breakdown, by controlling the accessibility of88 functional glycosaminoglycan fragments (Wan et al., 2020). By89 interacting with receptors located on the cell surface, like integrins90 and growth factor receptors, GAG fragments can encourage91 cytoskeleton remodeling and improve cell motility (Casale &92 Crane, 2024).93 The residues of breakdown may change the extracellular94 matrix’s mechanical features and the substance, which may95 affect how cells engage with their surroundings (Wang et al.,96 2017). Pattern recognition receptors (PRRs) on immune cell97 populations identify particular glycosaminoglycan components98 as danger-associated molecular patterns (DAMPs), which then,99 in turn, cause inflammatory reactions (Garantziotis & Savani,100 2022). About carcinoma, the breakdown components of101 glycosaminoglycan might affect the activity of the cancerous cells 102 and the stromal cells that envelop them, therefore facilitating the 103 growth, blood vessel development, and territorial expansion of 104 the tumor (Hua et al., 2022). Prospective treatment options for 105 recovering 𝛽-glucuronidase function in MPS VII patients include 106 gene therapy and enzyme replacement therapy (ERT) (Grubb 107 et al., 2010). Furthermore, the use of small-molecule chaperones 108 to regulate or increase the efficiency of mutant 𝛽-glucuronidase 109 has already been investigated (Doherty et al., 2023). 110 Single-nucleotide polymorphisms (SNPs), particularly non- 111 synonymous SNPs (nsSNPs), are major contributors to genetic 112 variability and are frequently implicated in inherited metabolic 113 disorders. Despite numerous reported variants, a systematic 114 approach to distinguish pathogenic mutations from benign 115 polymorphisms remains limited. Given the increasing volume of 116 genomic data generated through high-throughput sequencing, 117 there is a critical need to prioritize functionally significant 118 variants using reliable approaches. Identifying deleterious 119 nsSNPs and understanding their structural and functional 120 consequences can provide valuable insights into disease 121 mechanisms, genotype–phenotype correlations, and molecular 122 instability associated with protein dysfunction. Therefore, this 123 study was undertaken to comprehensively analyze nsSNPs in 124 the GUSB gene using integrative sequence- and structure-based 125 bioinformatics tools. By predicting pathogenicity, assessing 126 structural destabilization, and evaluating aggregation propensity, 127 the work aims to bridge the gap between genetic variation 128 data and mechanistic understanding. Ultimately, such insights 129 may facilitate improved diagnostic interpretation, biomarker 130 identification, and the development of targeted therapeutic 131 interventions.
- Primary disease focus: MPS VII (Sly syndrome)
- Target protein: β-glucuronidase
- Target gene: GUSB
- Protein length mentioned: 651 amino acids
- Core biological context: lysosomal glycosaminoglycan degradation and protein instability caused by missense variants
3. Materials and Methods
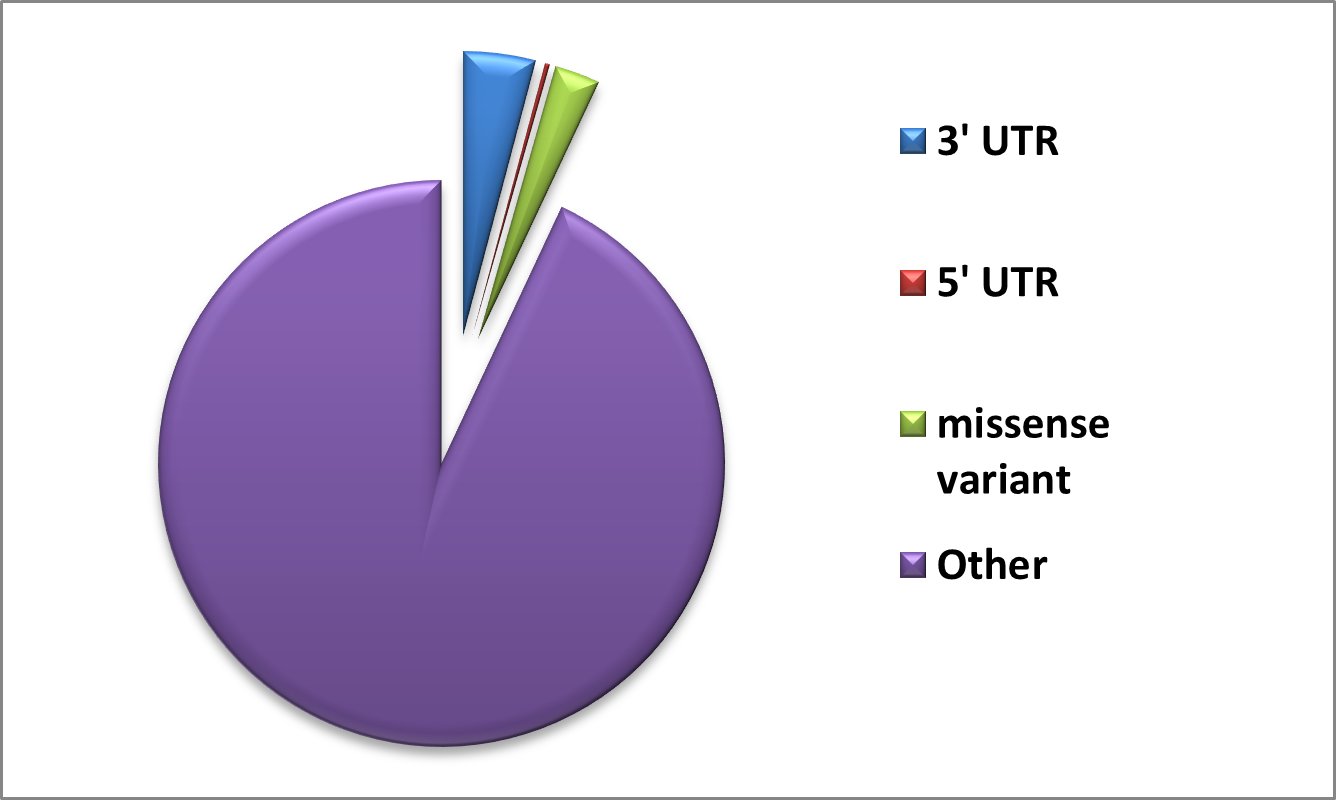
2.1. Data retrieval and analysis 134 The GUSB sequence was extracted in FASTA format from the 135 UniProt repository (UniProt ID: P08236). A collection of non- 136 synonymous SNPs was generated using information obtained 137 from the PubMed literature search and databases such as dbSNP 138 (Sherry et al., 2001), HGMD (Stenson et al., 2009), ClinVar 139 (Landrum et al., 2014), and Ensembl (Hubbard et al., 2002). The 140 list was cleared to eliminate redundant nsSNPs. The protein data 141 bank provided the crystal structure of the human GUSB gene. Out 142 of the total Data, 1305 Missense variants along with 3’UTR and 5’ 143 UTR were obtained from dbSNP and Ensembl, and are shown in 144 Figure 1. We used a comprehensive computational approach to 145 anticipate harmful mutations in the GUSB at both structural and 146 functional levels, as illustrated in Figure 2. 147
2.2. Sorting Intolerant from Tolerant 148 Sorting Intolerant from Tolerant (SIFT) was used to predict 149 how possible amino acid alterations may affect protein function. 150 SIFT is expanded to include predictions for frame-shifting indels. 151 Human genetic research has extensively used SIFT to assess amino 152 acid substitutions (e.g., cancer, Mendelian disorders, and viral 153 infections). Studies of human diseases and other study subjects 154 are not the only areas in which SIFT is useful. The consequences 155 of missense mutations on model animals such as rats, dogs, and 156 Arabidopsis, as well as agricultural plants, have been studied using 157 SIFT (Sim et al., 2012). If the SIFT score is less than or equal to 158 0.05, then the mutation is not tolerable (Ng & Henikoff, 2003). The 159 SIFT method was utilized to forecast the impact of these nsSNPs 160 on the protein.
2.3. Polymorphism Phenotyping v2 163 Polymorphism Phenotyping v2 (PolyPhen-2), a program and 164 Web server accessible, estimates the potential effects of amino 165 acid alterations on the stability and activity of human proteins. 166 SNPs are functionally annotated; coding SNPs are mapped to 167 genetranscripts;proteinsequenceannotationsareextracted;and 168 conservation profiles are constructed. The likelihood that the 169 missensemutationwouldcauseharmisthencalculatedusinga 170 combinationofallthesecharacteristics(Adzhubeietal.,2013). In additiontoestimatingthelikelihoodthatamutationisharmful171 172 when it is not, PolyPhen-2 also provides predictions of false- 173 positiveandtrue-positiverates. PolyPhen-2computesthenaive 174 Bayesposteriorprobabilitythataparticularmutationisharmful. 175 Thequalitativeassessmentofamutationdetermineswhetherit 176 is benign, perhaps harmful, or likely harmful (Adzhubei et al., 177 2010). 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 2.4. Mutation Assessor Mutation Assessor (http://mutationassessor.org/r3/) is a sequence-based tool that predicts the functional impact of an nsSNPsonaprotein. Thisserverpredictsthefunctionaleffectsof missensepolymorphismsandaminoacidmodificationsobserved in cancer-causing mutations in proteins. Depending on the affectedaminoacidsandthedegreeofevolutionarypreservation in protein homologs, the functional impact is evaluated (?). A sizablecollectionofpolymorphicanddisease-associated(OMIM) variants—60,000intotalhasbeenusedtoverifythetechnique. 2.5. Function Analysis through Hidden Markov Models FunctionAnalysisthroughHiddenMarkovModels(FATHMM) isamethodthathelpspredicthowbothcodingandnon-coding variationsinthehumangenomeaffectfunction. Todetermineif SNPsandothermutationstendtobedamaging,hiddenMarkov models are employed. When it comes to genetic research and personalizedtherapy,FATHMMishighlybeneficial,asithelpsto clarifythesignificanceofgeneticvariants(Shihabetal.,2013). 195 2.6. Mutation Cutoff Scanning Matrix 196 A computer method called Mutation Cutoff Scanning Matrix 197 (mCSM) is used to predict how mutations may affect small- 198 moleculebinding,protein-proteininteractions,protein-nucleic 199 acidinteractions, andproteinstability. mCSMisaninnovative 200 tool for assessing non-synonymous mutations that predicts 201 destabilizing mutations using a graph-based method. If a 202 mutation results in a mCSM score (∆∆𝐺) less than 0, it affects 203 proteinstructure(Choudhuryetal.,2021). Itassistsinforecasting 204 theimpactofmutationsondrugbinding,hencecontributingto 205 thedevelopmentofmorepotentmedications. Ithelpsunderstand 206 how changes in protein interactions and stabilization lead to 207 conditionscausedbymutations. Directsthecreationofproteins 208 foruseinindustryandmedicinethathavetheappropriatestability 209 andinteractioncharacteristics(Piresetal.,2016). 210 2.7. MAESTRO 211 Thefreeenergychangeonproteinunfoldingisestimatedusing212 MAESTRO(https://pbwww.che.sbg.ac.at/maestro/web),amulti- 213 agentstabilitypredictiontool. Itdetermineshowapointmutation214 affectstheprotein’sstabilitybycalculatingthedifferenceinfree 215 energy (∆𝐺) between the mutant and wild-type proteins. The 216 stability of the protein is changed by a mutation if its score is217 less than zero (Laimer et al., 2015). Mutations may be limited 218 to certain amino acid classes, exposed or buried residues, and 219 user-specifiedareas,contingentuponthepurposeoftheresearch 220 (Laimeretal.,2015). 221 2.8. PREMPS 222 PremPScalculateschangesintheunfoldingGibbsfreeenergyto 223 assess the effects of single mutations on protein stability. This224 approachrequiresaprotein’sthree-dimensionalstructure(Chen 225 et al., 2020). The PREMPS software determines the mutation’s 226 correspondingchangeinfreeenergy(∆∆𝐺). PREMPSforecasts 227 the impact of the mutation on protein stability based on the 228 calculated∆∆𝐺. Generally,adestabilizingmutationisindicated 229 byahigh∆∆𝐺value,whereasastabilizingmutationissuggested 230 byanegative∆∆𝐺(Chenetal.,2020). 231 2.9. DynaMut 232 DynaMutforecaststheeffectsofmutationsonproteinstabilityby 233 consideringMDsimulationoutcomes. Theproteinstructureis 234 stabilizedordestabilizedbyamutationbasedonthecalculation 235 ofmetricssuchaschangesinfreeenergy(∆∆𝐺). Thisprediction 236 is essential for evaluating how mutations in biological systems 237 will affect functionality (Rodrigues et al., 2018). PhD-SNP is a 238 tooldesignedtohelpunderstandthegeneticbasisofillnesses. It 239 analyzesSNPsbyassessingtheirpossiblenegativeconsequences 240 usingavarietyofstructuralandevolutionaryfactors. Aprediction241 scorethatindicatestheprobabilityofanSNPbeingdeleteriousis242 producedbyPhD-SNP.Agreaterscoredenotesahigherlikelihood 243 thattheSNPwouldresultinfunctionalalterationsorincreased 244 susceptibilitytoillness,withascorerangefrom0to1(Capriotti 245 etal.,2006). 246 2.10. SNP & GO 247 A bioinformatics technique called SNP & GO (http://snps-a 248 nd-go.biocomp.unibo.it/snps-and-go/) integrates data from 249 both Gene Ontology (GO) and SNP annotations to forecast the 250 functionaleffectsofSNPs. Thistoolfacilitatesunderstandingof 251 genomicdatainrelationtodiseaseandphenotypebyilluminating 252 thepotentialeffectsofgeneticvariantsonproteinfunctionand 253 3 Elucidating impacts of deleterious Missense mutations on the structure and function of 𝛽-glucuronidase: Investigating the molecular basis of pathogenesis of MPSVII 254 biological mechanisms. The algorithms used here categorize 255 SNPsasneutralordeleterious(likelytoimpairproteinfunction) 256 basedontheattributesthatwereretrieved. SNP&GOproduces 257 predictionscoresdeterminedbythemachinelearningmodelthat 258 showhowlikelyitisthatanSNPwillhavenegativeeffects. Higher 259 scoresindicateagreaterlikelihoodoffunctionalimpact,which 260 helpsresearcherschooseSNPsforfurtherstudy(Capriottietal., 261 2013). 262 2.11. MutPred2 263 MutPred2 was created to predict how missense mutations in 264 humanproteinsmightaffectproteinfunction. Itevaluateshow 265 mutationsinfluenceproteinstructure,function,andinteractions 266 by combining diverse features and machine learning methods, 267 deliveringsignificantinsightsintotheirpotentialrolesindisease 268 processes(Choudhuryetal.,2021). 269 2.12. Solubility based on Disorder and Aggregation 270 A technique to determine the aggregation, disorder, helix, and strandpropensitiesresultingfrommutationsiscalledSolubility271 272 based on Disorder and Aggregation (SODA) (http://protein. 273 bio.unipd.it/soda/). The PDB format structure file or the protein sequence may be entered into this program. SODA274 275 uses Fells, ESpritz-NMR, PASTA 2.0, and other resources to 276 forecastmutationsofmanytypes,includinginsertions,deletions, 277 substitutions, and duplications. Based on how differently the 278 WTandmutantproteinsaresoluble,SODAassignsafinalscore 279 (Paladinetal.,2017). 280 2.13. Consurf Analysis 281 Consurf (https://consurf.tau.ac.il/) is a bioinformatics tool 282 designed to find areas in protein sequences that have been 283 conservedacrosstime. Itusesevolutionarylinksamongvarious 284 species and sequence homology to predict the structural and 285 functional significance of amino acid residues. Using multiple 286 sequencealignment,theConSurftoolwasusedtoassessresidue 287 conservationataparticularlocation(Ashkenazyetal.,2016).
- UniProt ID: P08236
- Databases mentioned: PubMed, dbSNP, HGMD, ClinVar, Ensembl, UniProt, Protein Data Bank
- Sequence-based tools: SIFT, PolyPhen-2, Mutation Assessor, FATHMM
- Structure-based tools: mCSM, MAESTROweb, PremPS, DynaMut
- Pathogenicity tools: PhD-SNP, SNPs&GO, MutPred2
- Additional analysis tools: SODA, ConSurf
2.1. Data retrieval and analysis
The GUSB FASTA sequence was taken from UniProt, and a non-redundant list of nsSNPs was assembled from public databases and literature.
The text also states that dbSNP and Ensembl contained 1305 variants including missense, 3' UTR, and 5' UTR categories, visualized in Figure 1.
- Sequence source: UniProt
- Variant sources: dbSNP, HGMD, ClinVar, Ensembl, PubMed
- Visualization of overall variant categories: Figure 1
2.2–2.5. Sequence-based tools
SIFT, PolyPhen-2, Mutation Assessor, and FATHMM were used to predict the functional impact of amino acid substitutions.
These tools rely on conservation, structural features, physicochemical change, and hidden Markov model–based reasoning to classify tolerated versus damaging or low versus high impact substitutions.
- SIFT deleterious threshold described: score ≤ 0.05
- PolyPhen-2 qualitative classes: benign, possibly harmful, likely harmful
- Mutation Assessor purpose: functional impact estimation
- FATHMM purpose: functional prediction using hidden Markov models
2.6–2.9. Structure-based tools
mCSM, MAESTRO, PREMPS, and DynaMut were used to estimate mutation-induced effects on stability and conformational dynamics.
These approaches rely on ∆∆G or related stability metrics, graph-based signatures, energetic models, and simulation-aware estimates.
- mCSM: destabilizing if ∆∆G < 0
- MAESTRO: estimates change in unfolding free energy
- PREMPS: assesses single-mutation effects on unfolding Gibbs free energy
- DynaMut: estimates mutation effects on stability and conformational dynamics
2.10–2.13. Pathogenicity, solubility, and conservation analysis
SNPs&GO and MutPred2 were used alongside PhD-SNP to prioritize disease-associated variants after deleteriousness screening.
SODA was used to study aggregation and solubility changes, while ConSurf was applied to map evolutionary conservation across the GUSB protein.
- Pathogenicity platforms: SNPs&GO, MutPred2, PhD-SNP
- Aggregation/solubility platform: SODA
- Conservation platform: ConSurf
4. Results and Discussion
The results section combines large-scale variant collection, sequence-based filtering, structure-based destabilization analysis, disease-phenotype prediction, conserved-residue mapping, SODA solubility analysis, and focused structural comparison of specific pathogenic substitutions.
The article emphasizes a stringent multi-tool strategy to identify variants that are consistently deleterious across both functional and structural predictors.
- Reported total missense variants discussed in results: 1305 initially found, 449 analyzed as nsSNPs
- High-confidence deleterious and destabilizing mutations: 15
- High-confidence pathogenic mutations prioritized: 8
- Key structurally highlighted residues: 566 and 388
3.1. Sequence-based predictions
The article states that 449 GUSB nsSNPs were analyzed using SIFT, PolyPhen-2, FATHMM, and Mutation Assessor.
The numbers reported as deleterious were 277 by SIFT, 262 by PolyPhen-2, 160 by FATHMM, and 192 by Mutation Assessor.
- SIFT deleterious calls: 277
- PolyPhen-2 deleterious calls: 262
- FATHMM deleterious calls: 160
- Mutation Assessor deleterious calls: 192
3.2. Structure-based prediction
The structure-based stage used DynaMut, PremPS, mCSM, and MAESTROweb to estimate mutation-induced destabilization and conformational effects.
The article reports 13 destabilizing mutations by mCSM, 40 by MAESTROweb, 1 by PremPS, and 11 by DynaMut, and then narrows these to 15 variants consistently selected by all sequence- and structure-based tools.
- mCSM destabilizing calls: 13
- MAESTROweb destabilizing calls: 40
- PremPS destabilizing calls: 1
- DynaMut destabilizing calls: 11
- Consensus deleterious and destabilizing variants retained: 15
3.3. Identification of pathogenic nsSNPs
The 15 prioritized variants were evaluated with PhD-SNP, SNPs&GO, and MutPred2 to classify likely disease-causing mutations.
Eight variants were highlighted as the strongest pathogenic candidates by overlap between the disease-prediction tools: L214P, L566P, N135D, P108L, R36L, T180P, Y388C, and Y399C.
- PhD-SNP disease predictions: 12 of 15
- SNPs&GO disease predictions: 10 of 15
- Overlap-prioritized pathogenic variants: L214P, L566P, N135D, P108L, R36L, T180P, Y388C, Y399C
3.4. Analysis of evolutionarily conserved residues
ConSurf analysis identified strongly conserved stretches around residues 360–370, 380–390, 415–425, and 566–571.
The text explicitly notes that L566P and Y388C are located in highly conserved regions, increasing confidence that these substitutions are structurally and functionally important.
- Highly conserved regions highlighted: 360–370, 380–390, 415–425, 566–571
- Pathogenic mutations explicitly tied to conserved regions: L566P, Y388C
3.5. Analysis of aggregation propensity
SODA analysis was performed on the eight disease-associated variants to evaluate solubility and aggregation behavior.
Three variants, R36L, Y388C, and Y399C, were predicted as less soluble, while the remaining five were predicted as more soluble.
- Total disease-associated variants tested by SODA: 8
- Less soluble variants: R36L, Y388C, Y399C
- More soluble variants: L214P, L566P, N135D, P108L, T180P
- Article's stated fraction with reduced solubility/increased aggregation tendency: 37.5%
3.6. Analyzing Structures of Specific Mutations
Focused structural comparison was conducted for mutations at residues 566 and 388.
For both positions, the mutant structures retained fewer conventional hydrogen bonds than the wild type and showed complete loss of aromatic interactions together with a dramatic drop in hydrophobic interactions, supporting a destabilizing effect.
- Residue 566 wild type: Leucine; mutant: Proline
- Residue 388 wild type: Tyrosine; mutant: Cysteine
- Wild-type aromatic interactions reported: 8
- Mutant aromatic interactions reported: 0
- Wild-type hydrophobic interactions reported: 17
- Mutant hydrophobic interactions reported: 1
5. Conclusion
The paper concludes that integrative computational analysis can prioritize disease-relevant GUSB variants by combining sequence-level deleteriousness prediction, structure-based destabilization analysis, pathogenicity scoring, conservation mapping, and solubility analysis.
It proposes that the findings could support future therapeutic strategies such as pharmacological chaperones, solubility-enhancing agents, or other precision approaches aimed at restoring mutant β-glucuronidase function.
- Overall prioritized pathogenic variants: 8
- Mechanistic emphasis: structural destabilization, altered interactions, and aggregation tendency
- Future direction: experimental validation and therapeutic development
6. Statements and Declarations
The article closes with declarations regarding conflicts of interest, funding, data availability, and use of AI tools.
- Conflicts of Interest: The authors declare no conflict of interest.
- Funding: This work received no funding.
- Data Availability Statement: All data generated or analyzed during this study are included in this manuscript.
- Declaration on the Use of AI Tools: The authors declare that ChatGPT (OpenAI) was used solely to refine the language, improve grammar, and enhance the clarity of the manuscript.
Figures
The PDF includes six figures covering overall SNP distribution, the computational analysis workflow, sequence-based and structure-based prediction summaries, conserved-residue mapping, and structural illustrations for specific prioritized mutations.
Figure 1

{kind=link}
Figure 1. Number of SNPs in GusB represented using the dbSNP database.
Download figure
Figure 2. A recap of the computational approaches used to anticipate harmful mutations in the GUSB at both structural and functional levels.
Download figure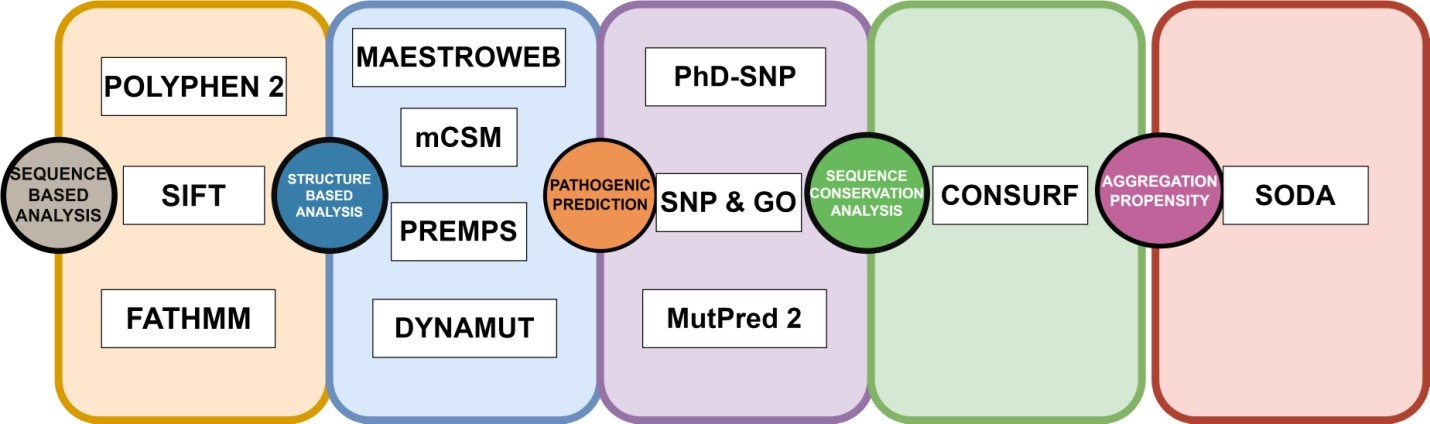{kind=link}

Figure 3. Distribution of deleterious and neutral nsSNPs predicted by sequence-based tools for the entire sequence of GUSB gene.
Download figure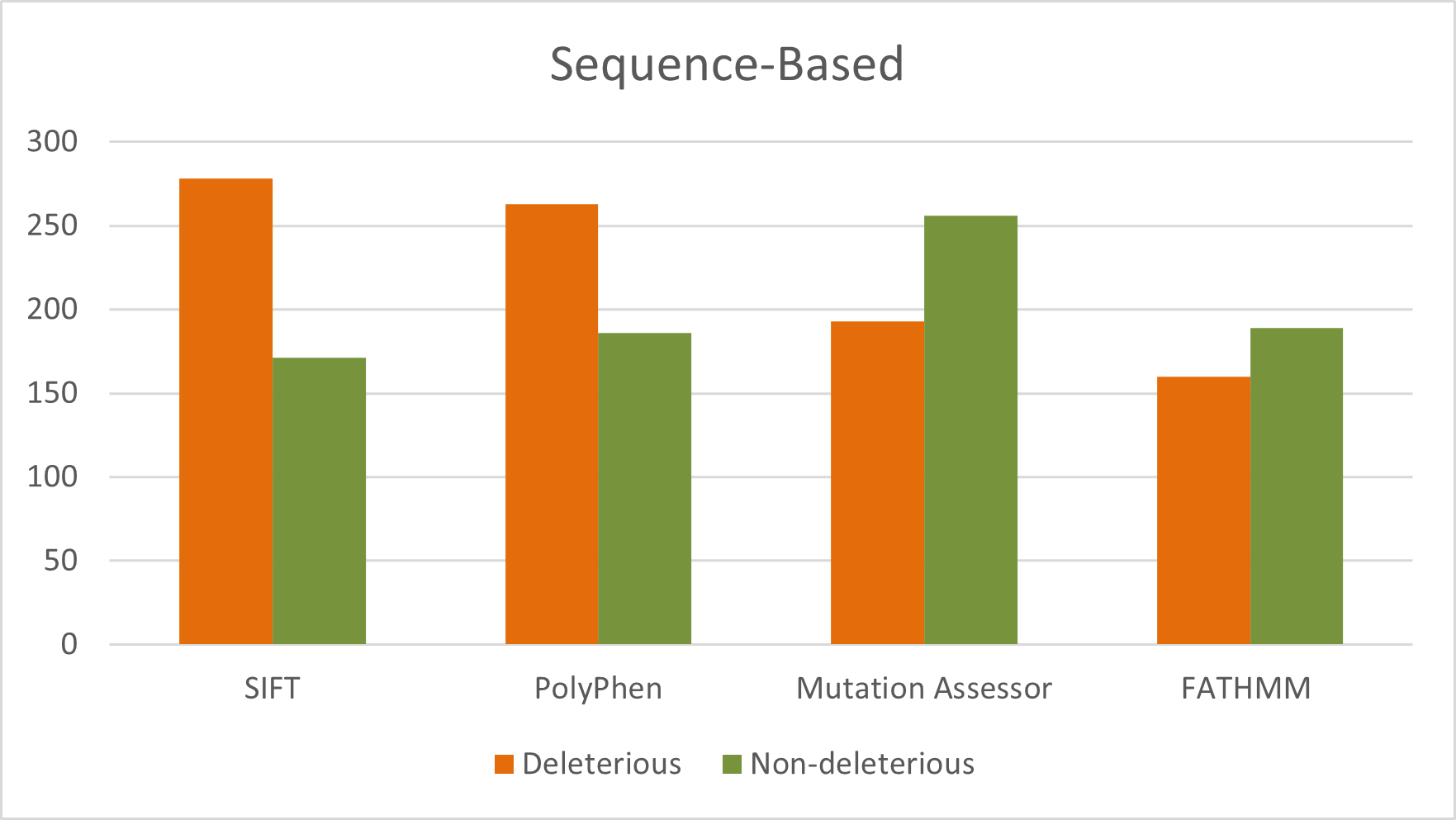{kind=link}

Figure 4. Distribution of destabilizing nsSNPs predicted by structure-based tools for the entire sequence of GUSB gene.
Download figure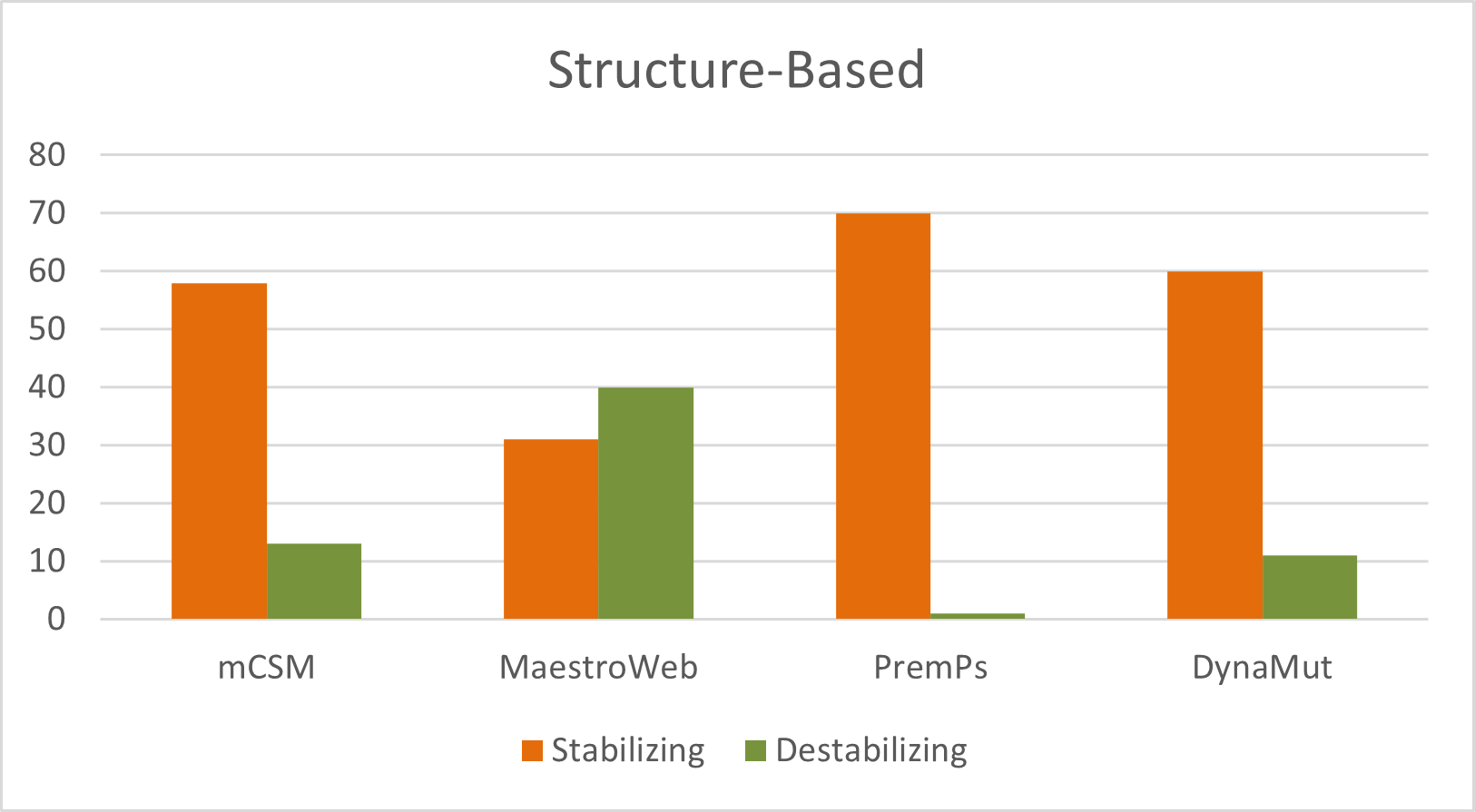{kind=link}
Figure 5. Conserved region of GusB.

Figure 6. Analysis of specific mutations: (A) leucine residue 566 mutated to proline and (B) tyrosine to cysteine at position 388.
Download figure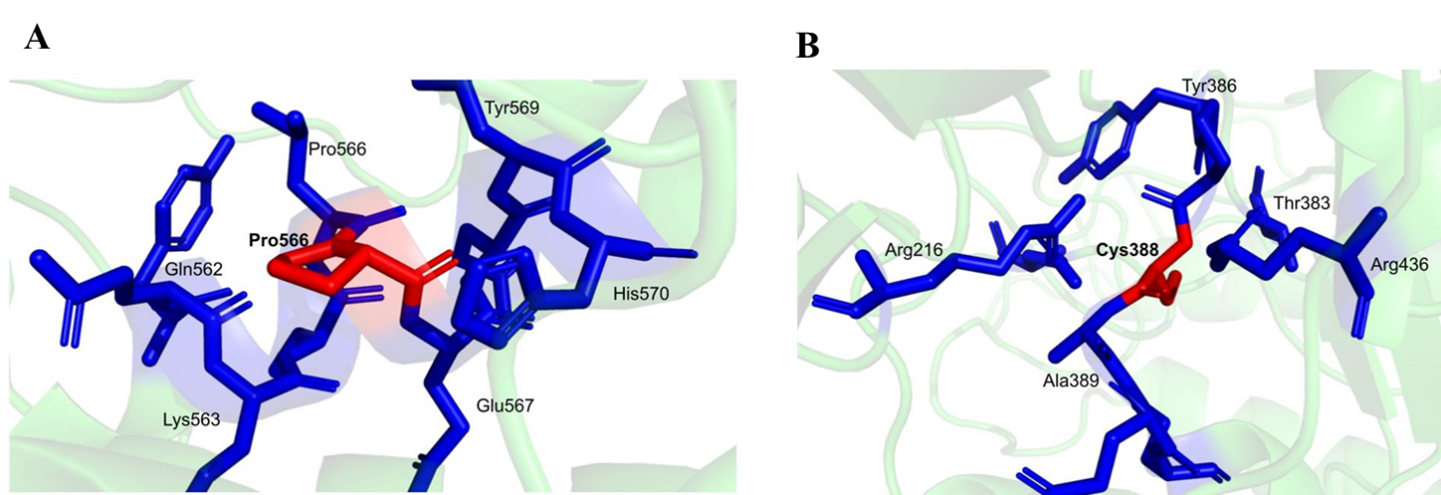{kind=link}
References
- 1
Adzhubei, I., Jordan, D., & Sunyaev, S.
Current Protocols in Human Genetics article cited in the paper.
Current Protocols in Human Genetics · 2013 - 2
Adzhubei, I., Schmidt, S., Peshkin, L., et al.
Nature Methods article cited in the paper.
Nature Methods · 2010 - 3
Ashkenazy, H., Abadi, S., Martz, E., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2016 - 4
Awolade, P., Cele, N., Kerru, N., et al.
European Journal of Medicinal Chemistry article cited in the paper.
European Journal of Medicinal Chemistry · 2020 - 5
Balch, W., Morimoto, R., Dillin, A., & Kelly, J.
Science article cited in the paper.
Science · 2008 - 6
Capriotti, E., Calabrese, R., & Casadio, R.
Bioinformatics article cited in the paper.
Bioinformatics · 2006 - 7
Capriotti, E., Calabrese, R., Fariselli, P., et al.
BMC Genomics article cited in the paper.
BMC Genomics · 2013 - 8
Casale, J., & Crane, J.
StatPearls entry cited in the paper.
StatPearls · 2024 - 9
Chen, Y., Lu, H., Zhang, N., et al.
PLoS Computational Biology article cited in the paper.
PLoS Computational Biology · 2020 - 10
Choudhury, A., Mohammad, T., Samarth, N., et al.
Scientific Reports article cited in the paper.
Scientific Reports · 2021 - 11
Doherty, G., Ler, G., Wimmer, N., et al.
ChemBioChem article cited in the paper.
ChemBioChem · 2023 - 12
Florindo, R., Souza, V., Mutti, H., et al.
New Biotechnology article cited in the paper.
New Biotechnology · 2018 - 13
Garantziotis, S., & Savani, R.
American Journal of Physiology - Cell Physiology article cited in the paper.
American Journal of Physiology - Cell Physiology · 2022 - 14
Grubb, J., Vogler, C., & Sly, W.
Rejuvenation Research article cited in the paper.
Rejuvenation Research · 2010 - 15
Hua, S., Viera, M., Yip, G., & Bay, B.
Cancers article cited in the paper.
Cancers · 2022 - 16
Hubbard, T., Barker, D., Birney, E., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2002 - 17
Hytönen, M., Arumilli, M., Lappalainen, A., et al.
PLoS ONE article cited in the paper.
PLoS ONE · 2012 - 18
Khan, S., Peracha, H., Ballhausen, D., et al.
Molecular Genetics and Metabolism article cited in the paper.
Molecular Genetics and Metabolism · 2017 - 19
Knowles, T., Vendruscolo, M., & Dobson, C.
Nature Reviews Molecular Cell Biology article cited in the paper.
Nature Reviews Molecular Cell Biology · 2014 - 20
Kong, X., Zheng, Z., Song, G., et al.
Frontiers in Immunology article cited in the paper.
Frontiers in Immunology · 2022 - 21
Laimer, J., Hofer, H., Fritz, M., Wegenkittl, S., & Lackner, P.
BMC Bioinformatics article cited in the paper.
BMC Bioinformatics · 2015 - 22
Landrum, M., Lee, J., Riley, G., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2014 - 23
Nagpal, R., Goyal, R., Priyadarshini, K., et al.
Indian Journal of Ophthalmology article cited in the paper.
Indian Journal of Ophthalmology · 2022 - 24
Naz, H., Islam, A., Waheed, A., et al.
Rejuvenation Research article cited in the paper.
Rejuvenation Research · 2013 - 25
Ng, P., & Henikoff, S.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2003 - 26
Paladin, L., Piovesan, D., & Tosatto, S.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2017 - 27
Pires, D., Blundell, T., & Ascher, D.
Scientific Reports article cited in the paper.
Scientific Reports · 2016 - 28
Rodrigues, C., Pires, D., & Ascher, D.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2018 - 29
Sawamoto, K., Álvarez González, J., Piechnik, M., et al.
International Journal of Molecular Sciences article cited in the paper.
International Journal of Molecular Sciences · 2020 - 30
Seker Yilmaz, B., Davison, J., Jones, S., & Baruteau, J.
Journal of Inherited Metabolic Disease article cited in the paper.
Journal of Inherited Metabolic Disease · 2021 - 31
Sherry, S., Ward, M., Kholodov, M., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2001 - 32
Shihab, H., Gough, J., Cooper, D., et al.
Human Mutation article cited in the paper.
Human Mutation · 2013 - 33
Shihab, H., Rogers, M., Gough, J., et al.
Bioinformatics article cited in the paper.
Bioinformatics · 2015 - 34
Sim, N.-L., Kumar, P., Hu, J., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2012 - 35
Staudt, C., Puissant, E., & Boonen, M.
International Journal of Molecular Sciences article cited in the paper.
International Journal of Molecular Sciences · 2016 - 36
Stenson, P., Mort, M., Ball, E., et al.
Genome Medicine article cited in the paper.
Genome Medicine · 2009 - 37
Thal, D., Walter, J., Saido, T., & Fändrich, M.
Acta Neuropathologica article cited in the paper.
Acta Neuropathologica · 2015 - 38
Tomatsu, S., Montaño, A., Oikawa, H., et al.
Current Pharmaceutical Biotechnology article cited in the paper.
Current Pharmaceutical Biotechnology · 2011 - 39
Urayama, A., Grubb, J., Sly, W., & Banks, W.
Proceedings of the National Academy of Sciences of the United States of America article cited in the paper.
Proceedings of the National Academy of Sciences of the United States of America · 2004 - 40
Wan, L., Zhang, S., Li, S., et al.
Thrombosis and Haemostasis article cited in the paper.
Thrombosis and Haemostasis · 2020 - 41
Wang, M., Liu, X., Lyu, Z., et al.
Colloids and Surfaces B: Biointerfaces article cited in the paper.
Colloids and Surfaces B: Biointerfaces · 2017 - 42
Yang, G., Ge, S., Singh, R., et al.
Drug Metabolism Reviews article cited in the paper.
Drug Metabolism Reviews · 2017
Tables
Table 1. Disease phenotype analysis of high-confidence nsSNPs in the GUSB gene.
| S. No. | Mutations | PhD-SNP | SNP & GO | MutPred2 |
|---|---|---|---|---|
| 1 | I499M | Neutral | Neutral | 0.62 |
| 2 | L121V | Neutral | Neutral | 0.77 |
| 3 | L214P | Disease | Disease | 0.953 |
| 4 | L566P | Disease | Disease | 0.888 |
| 5 | N135D | Disease | Disease | 0.815 |
| 6 | P108L | Disease | Disease | 0.8 |
| 7 | P196H | Neutral | Disease | 0.701 |
| 8 | R36L | Disease | Disease | 0.943 |
| 9 | R398H | Disease | Neutral | 0.561 |
| 10 | R56C | Disease | Neutral | 0.361 |
| 11 | T180P | Disease | Disease | 0.843 |
| 12 | T226P | Disease | Disease | 0.497 |
| 13 | V601G | Disease | Neutral | 0.545 |
| 14 | Y388C | Disease | Disease | 0.785 |
| 15 | Y399C | Disease | Disease | 0.567 |
Table 2. Prediction of aggregation propensity of mutant GUSB using SODA server.
| Mutations | Sequence | Helix | Strand | Aggregation | Disorder | SODA | Result |
|---|---|---|---|---|---|---|---|
| Wild type | 0.281 | 0.296 | -5.254 | 0.024 | |||
| L214P | 1 | -1.351 | 0.165 | 18.495 | 0.024 | 16.507 | More soluble |
| L566P | 2 | -2.1 | 0.568 | 5.083 | 0.034 | 2.178 | More soluble |
| N135D | 3 | 2.394 | -2.928 | 116 | 0.003 | 115.555 | More soluble |
| P108L | 4 | 0.707 | -0.875 | 12.252 | 0.004 | 11.977 | More soluble |
| R36L | 5 | -2.585 | 2.598 | -0.668 | -0.445 | -2.167 | Less soluble |
| T180P | 6 | -0.248 | -0.335 | 17.006 | 0.149 | 16.553 | More soluble |
| Y388C | 7 | -1.912 | 1.187 | -0.577 | -0.033 | -1.992 | Less soluble |
| Y399C | 8 | 0.028 | 0.82 | -22.711 | -0.069 | -21.467 | Less soluble |