


About Article
Identification of Potent Inhibitors of Ephrin type-A receptor 1 using Virtual Screening and Molecular Dynamics Simulation Approach
Volume 1, No. 1 · 2026
Highlights
- EphA1 was studied as a receptor tyrosine kinase target relevant to tumor progression, angiogenesis, metastasis, and immune regulation.
- A natural-compound virtual screen started from 90,000 ZINC molecules and was filtered to 32,901 drug-like compounds using Lipinski criteria.
- Docking shortlisted 320 compounds with binding energies from −11.2 to −9.4 kcal/mol.
- ZINC12660859 and ZINC12661003 emerged as the most promising post-filter candidates after PAINS, physicochemical, ADMET, carcinogenicity, and PASS analysis.
- ZINC12660859 showed stable binding in a 100 ns molecular-dynamics simulation with mean RMSD 0.26 nm, Rg 1.93 nm, SASA 139.62 nm², and 190 hydrogen bonds.
Abstract
Ephrin type-A receptor 1 (EphA1) is a receptor tyrosine kinase implicated in tumor progression, angiogenesis, metastasis, and immune regulation, making it an attractive therapeutic target in cancer.
A structure-based virtual screening approach was used to identify potential natural inhibitors of EphA1 from the ZINC database. From 90,000 natural compounds, 32,901 drug-like molecules passed Lipinski filtering and were docked against EphA1.
The three-dimensional target structure was taken from AlphaFold, docking was performed with InstaDock, and top hits were refined by PAINS filtering, physicochemical analysis, ADMET profiling, carcinogenicity prediction, PASS analysis, and protein–ligand interaction analysis.
Two compounds, ZINC12660859 and ZINC12661003, showed favorable drug-likeness, high gastrointestinal absorption, non-carcinogenicity, and predicted antineoplastic activity.
Molecular-dynamics simulation of the EphA1–ZINC12660859 complex over 100 ns supported stable binding and suggested that ZINC12660859 is a promising computational lead for EphA1-targeted anticancer discovery.
Keywords
Article Overview
This article presents an integrative structure-based computational workflow for discovery of EphA1 inhibitors, motivated by EphA1’s role in cancer, angiogenesis, metastasis, immune signaling, and neuroinflammation.
The paper explains EphA1 biology, outlines why the ATP-binding pocket of the kinase domain is an appealing therapeutic site, and then applies screening, docking, pharmacokinetic filtering, biological-activity prediction, and molecular-dynamics simulation to prioritize lead compounds.
The final emphasis of the article is not simply on docking score ranking, but on identifying a computationally credible lead that combines acceptable drug-likeness with stable dynamic behavior in the protein pocket.
1. About the Article
This is a research article published in Clinical & Molecular Biomedicine. It reports a computational discovery workflow for potential EphA1 inhibitors using natural compounds from the ZINC database.
The work involves researchers affiliated with Jamia Millia Islamia, Cleveland Clinic, and Case Western Reserve University. Nida Mubin is listed as the corresponding author.
- Journal: Clinical & Molecular Biomedicine
- Volume/Issue: Vol. 1, No. 1
- Header year: 2026
- Title: Identification of Potent Inhibitors of Ephrin type-A receptor 1 using Virtual Screening and Molecular Dynamics Simulation Approach
- Authors: Shanza Rehman; Sana Kauser; Syed Suhail Andrabi; Nida Mubin
- Affiliation 1: Department of Computer Science, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India
- Affiliation 2: Department of Biosciences, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India
- Affiliation 3: Biomedical Engineering, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA
- Affiliation 4: Division of Haematology & Oncology, Case Western Reserve University, Cleveland, OH, USA
- Corresponding author: Nida Mubin
- Corresponding email: nxm793@case.edu
- Received: April 16th, 2024
- Accepted: March 31, 2026
- Published: May 21, 2024
- License: Creative Commons CC-BY 4.0
- Open-access status: Open Access
- Header page range on first page: pp. 1–25
- Printed article page range visible in PDF pages: 1–8
2. Introduction
The introduction frames Eph receptors as the largest receptor tyrosine kinase subgroup and explains that EphA1 participates in cell proliferation, differentiation, migration, survival, angiogenesis, immune regulation, and neuronal development.
The article summarizes disease relevance of EphA1 across cancers and other disorders. It states that EphA1 is overexpressed in thymoma, prostate cancer, hepatocellular carcinoma, ovarian cancer, breast cancer, clear cell renal cell carcinoma, gastric carcinoma, nasopharyngeal carcinoma, and oesophageal squamous cell carcinoma, while downregulated in colorectal cancer and uveal melanoma.
Because EphA1 contributes to tumor growth, angiogenesis, invasion, metastasis, and immune-related signaling, the ATP-binding pocket of its kinase domain is proposed as a rational inhibitory target.
The authors position structure-based drug design and virtual screening as efficient alternatives to trial-and-error discovery, then introduce an integrated workflow combining docking, pharmacokinetic filtering, PASS activity prediction, and molecular-dynamics simulation.
- Target protein: Ephrin type-A receptor 1 (EphA1)
- Gene: EPHA1
- Chromosomal location noted: 7q34
- Therapeutic rationale: inhibit ATP-binding pocket of kinase domain
- Therapeutic areas discussed: cancer, angiogenesis, neuroinflammation, Parkinson’s disease, Alzheimer’s disease, autoimmune and inflammatory disorders
3. Materials and Methods
The study used a Windows-based laptop with a 1.19 GHz CPU, 8 GB RAM, and a 512 GB SSD. InstaDock and Discovery Studio were used for screening and docking, PyMOL and Discovery Studio Visualizer for structural preparation and visualization, and GROMACS for molecular-dynamics simulation.
The EphA1 structure was selected from UniProt and AlphaFold, refined in PyMOL, and prepared by removing water molecules. The article identifies the screened receptor target as AF-P21709-F1.
A natural-compound library from the ZINC database was filtered with Lipinski’s rule of five, leaving 32,901 small molecules from an initial set of 90,000 compounds.
Docking used a defined grid centered at x = -1.511, y = 1.922, z = -35.295 with grid dimensions X = 56, Y = 63, Z = 75. The workflow then applied PAINS filtering, physicochemical filtering, ADMET analysis through pkCSM, carcinogenicity assessment through CarcinoPred-EL, and PASS activity prediction through Way2Drug PASS Online.
Molecular-dynamics simulations were performed for 100 ns at 300 K using GROMACS 5.1.2 with the G54a7 force field, a cubic water box, the spc216 water model, 1500 steepest-descent minimization steps, and standard trajectory analyses for RMSD, RMSF, Rg, SASA, and hydrogen bonding.
- Computing platform: Windows laptop, 1.19 GHz CPU, 8 GB RAM, 512 GB SSD
- Docking software: InstaDock
- Visualization software: PyMOL; Discovery Studio Visualizer
- Databases/resources: NCBI; UniProt; RCSB PDB; AlphaFold; ZINC; SwissADME; pkCSM; Way2Drug PASS; CarcinoPred-EL
- Simulation software: GROMACS 5.1.2
- Initial library size: 90,000 natural compounds
- Drug-like compounds after Lipinski filtering: 32,901
- Docking target identifier mentioned: AF-P21709-F1
- Docking grid center: (-1.511, 1.922, -35.295)
- Docking grid size: X=56, Y=63, Z=75
- Simulation length: 100 ns
- Simulation temperature: 300 K
- Force field: G54a7
- Water model: spc216
2.1. Web Resources and the Computing Environment
The article enumerates the computational environment and tools used throughout the study, emphasizing fully in silico screening, docking, profiling, and MD simulation.
The workflow figure is referenced as Figure 1, although the text contains a placeholder form ('Figure ??') in one methods sentence.
- Reliable power source and high-speed internet were explicitly noted.
- QtGrace was used for graph generation in MD analysis.
2.2. Receptor and Library Preparation
EphA1 was selected based on structural completeness, kinase-domain presence, mutation-related sequence information, ligand-related information, and structural purity.
The article includes a brief background note on ZINC and cites both its scale and update frequency before describing the final filtered subset used for docking.
- Structure source: AlphaFold Protein Structure Database
- Protein preparation step explicitly mentioned: water removal
- Figure 2 shows the prepared kinase domain
2.3. Molecular Docking and Virtual Screening
Ligands were obtained in pdbqt format and docked to examine bond conformations and binding affinity in the active-site cavity.
The workflow used random seed generation and scoring-based ranking, after which log files and output files were reviewed to choose suitable docked conformations.
- Post-docking visualization tools: PyMOL and Discovery Studio Visualizer
- Activity threshold statement in PASS section: activities with Pa greater than 0.71 were considered
2.4. Drug-Likeness, PAINS, and ADMET Analysis
SwissADME-based PAINS filtering was used to remove likely false positives, while total polar surface area and solubility predictions were applied as additional filters.
pkCSM was used for ADMET analysis, specifically assessing GI absorption, BBB permeability, CYP2D6 inhibition, OCT2 substrate status, and AMES toxicity.
- TPSA filter range: 65–145 Ų
2.5. PASS Analysis and Protein–Ligand Interaction Analysis
PASS analysis focused on predicted biological activities with special interest in antineoplastic potential.
Interaction analysis emphasized hydrogen bonding, hydrophobic contacts, van der Waals interactions, and especially interactions with Lys656 and Asp749 in the ATP-binding pocket.
2.6. MD simulations
MD simulations were performed both for apo EphA1 and for the EphA1–ZINC12660859 complex.
The trajectory analysis used GROMACS utilities for RMSD, RMSF, radius of gyration, SASA, and hydrogen-bond evaluation.
4. Results and Discussion
From 32,901 screened compounds, the authors report 320 compounds with notable binding affinities between −11.2 and −9.4 kcal/mol. Table 1, however, lists the top 66 compounds explicitly, with ZINC03845566 showing the strongest listed affinity (−11.2 kcal/mol).
After PAINS and physicochemical filtering, 24 compounds remained for ADMET analysis, and 20 reportedly passed ADMET-related criteria. PASS activity analysis then narrowed the focus to ZINC12660859 and ZINC12661003.
Interaction analysis placed both compounds in or near the ATP-binding site, with repeated emphasis on Lys656 and Asp749 as relevant residues. ZINC12660859 in particular formed hydrogen bonds with Asp749, Gly769, and Gly633 and multiple additional interactions with residues surrounding the kinase pocket.
Molecular-dynamics analysis focused on ZINC12660859 and concluded that the ligand-bound complex remained stable over 100 ns with only small changes in RMSD, RMSF, compactness, surface exposure, and hydrogen-bond counts.
- Screened docked set after Lipinski filtering: 32,901
- Compounds said to have significant binding affinity: 320
- Explicitly tabulated top compounds: 66
- Top listed docking score: ZINC03845566 at −11.2 kcal/mol
- Lead candidates after multi-step filtering: ZINC12660859 and ZINC12661003
- Final MD focus compound: ZINC12660859
3.1. Virtual Screening and Binding Affinity Analysis
The screening produced log files and docked poses for each ligand, from which compounds were ranked by binding affinity and pose quality.
The paper highlights ZINC03845566 (−11.2 kcal/mol), ZINC12660859 (−9.9 kcal/mol), and ZINC12661003 (−9.7 kcal/mol), while noting that the latter two were preferred after downstream pharmacokinetic and biological-activity filters.
- Binding-energy range highlighted for shortlisted compounds: −11.2 to −9.4 kcal/mol
- Top listed compound: ZINC03845566
- Lead candidate 1: ZINC12660859
- Lead candidate 2: ZINC12661003
3.2. Drug Likeness and ADMET Analysis
SMILES strings were generated in Discovery Studio and submitted to SwissADME for PAINS filtering. Compounds violating Lipinski rules, acceptable TPSA range, or solubility filters were removed.
The paper states that 24 compounds remained after filtering and that 20 successfully met ADMET parameters. Two compounds were then retained based on PASS-supported anti-cancer relevance.
- ZINC12660859: molecular weight 498.664, rotatable bonds 5, H-bond donors 2, H-bond acceptors 5, LogP 3.9952, PAINS 0
- ZINC12661003: molecular weight 404.459, rotatable bonds 0, H-bond donors 0, H-bond acceptors 7, LogP 2.6825, PAINS 0
- ZINC12660859 GI absorption: 94.952 (High)
- ZINC12661003 GI absorption: 99.476 (High)
- Both compounds: not BBB permeant, not CYP2D6 inhibitors, not OCT2 substrates, not AMES toxic according to Table 3
3.3. Interaction Analysis
The article reports 18 docked conformers for each of the two selected compounds and uses Discovery Studio to map binding residues and interaction types.
ZINC12660859 formed hydrogen bonds with Asp749, Gly769, and Gly633, while other interactions involved Lys656, Arg753, Asn754, Asp767, Val638, Ile630, Leu770, Leu756, Ala709, Gly631, Ala654, Phe704, Met705, Gly708, Phe635, and Glu634.
ZINC12661003 formed hydrogen bonds with Asp767 and Glu673 and other interactions with Lys656, Ser766, Leu686, Ala709, Gly708, Val638, Met705, Phe704, Ala654, Leu756, and Ile630.
- Key catalytic/ATP-pocket residues emphasized: Lys656; Asp749
- Figure 3 gives 2D interaction diagrams for both selected compounds.
- Figure 4 provides potential surface representations of both complexes.
3.4. MD Simulations
The MD section compares apo EphA1 with the EphA1–ZINC12660859 complex across RMSD, RMSF, radius of gyration, SASA, and hydrogen-bond analysis.
The mean RMSD values were 0.20 nm for EphA1 and 0.26 nm for the complex, and both systems approached equilibrium over the 100 ns trajectory.
RMSF was interpreted as lower in the ligand-bound system, suggesting reduced residual flexibility after binding, although the text contains one apparent mistaken reference to 'POT1 protein' instead of EphA1.
Mean radius of gyration changed only slightly from 1.92 nm to 1.93 nm, and mean SASA decreased from 140.42 nm² to 139.62 nm². Hydrogen-bond counts were 189 and 190, respectively, indicating overall maintenance of structural integrity.
- Apo EphA1 mean RMSD: 0.20 nm
- Complex mean RMSD: 0.26 nm
- Apo EphA1 mean RMSF: 0.13 nm
- Complex mean RMSF: 0.12 nm
- Apo EphA1 mean Rg: 1.92 nm
- Complex mean Rg: 1.93 nm
- Apo EphA1 mean SASA: 140.42 nm²
- Complex mean SASA: 139.62 nm²
- Apo EphA1 hydrogen bonds: 189
- Complex hydrogen bonds: 190
- Intermolecular hydrogen bonding described for ligand: commonly 1–2 stable H-bonds, with occasional 3–4
5. Conclusion
The paper concludes that ZINC12660859 is the most promising lead compound identified in the workflow because it combines acceptable docking, drug-likeness, ADMET behavior, predicted antineoplastic activity, and stable dynamic binding behavior in the EphA1 kinase pocket.
The authors emphasize that the work provides computational evidence only and explicitly call for in vitro and in vivo validation to confirm inhibitory activity and anticancer efficacy.
The study also presents structure-based virtual screening and molecular-dynamics simulation as an efficient framework for accelerating kinase-targeted therapeutic discovery.
- Preferred lead compound: ZINC12660859
- Target site: ATP-binding pocket of EphA1 kinase domain
- Validation still required: in vitro and in vivo
7. Statements and Declarations
The article contains closing statements for conflict of interest, data availability, funding, acknowledgements, supplementary materials, and author contributions.
- Conflict of Interest: There is no conflict of interest to declare.
- Data Availability Statement: The data supporting this study are provided in this article.
- Funding: None
- Acknowledgements: None
- Supplementary Materials: None
Figures
The PDF includes seven named figures illustrating the workflow, prepared receptor structure, ligand-interaction diagrams, surface views, and molecular-dynamics behavior of the EphA1 system.
Figure 1
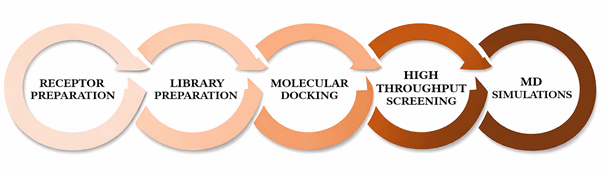
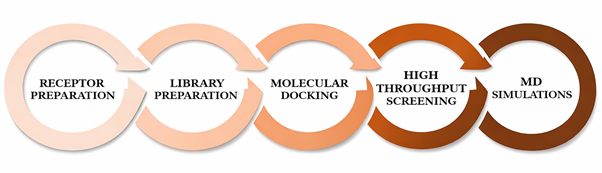{kind=link}
Figure 1. Steps involved in methodology.
Download figure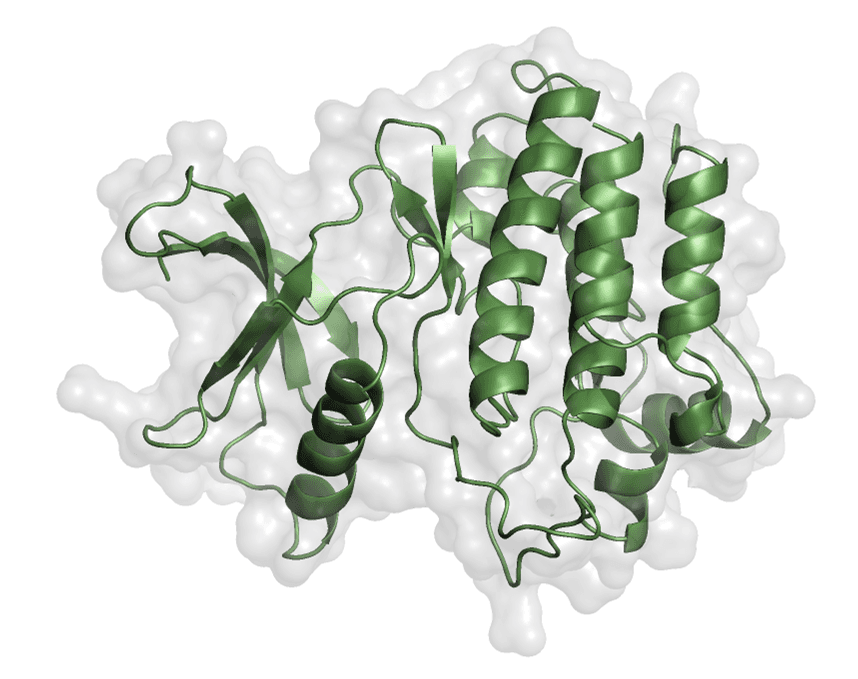
Figure 2. Prepared kinase domain of EphA1 shown in PyMOL.
Download figure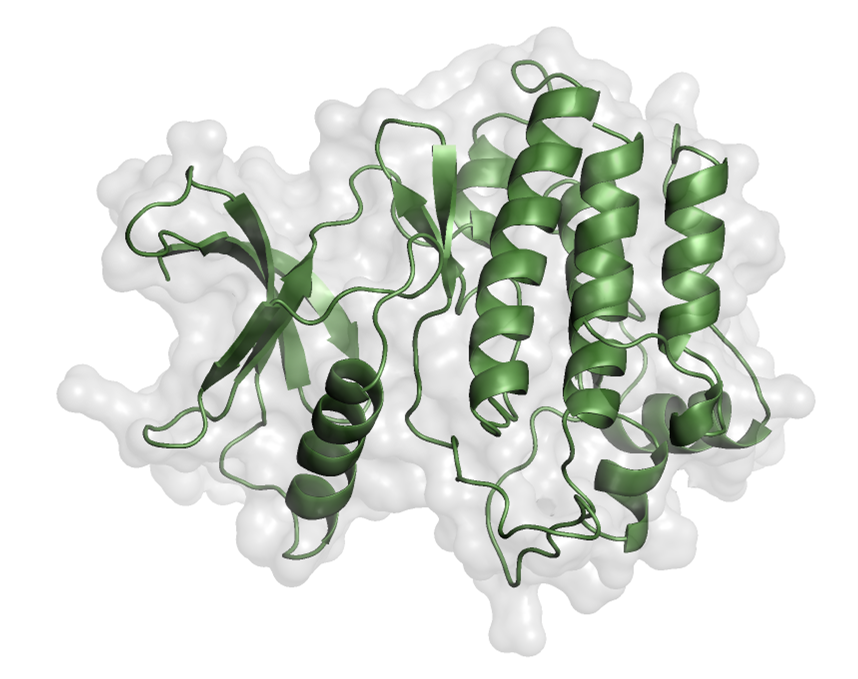{kind=link}
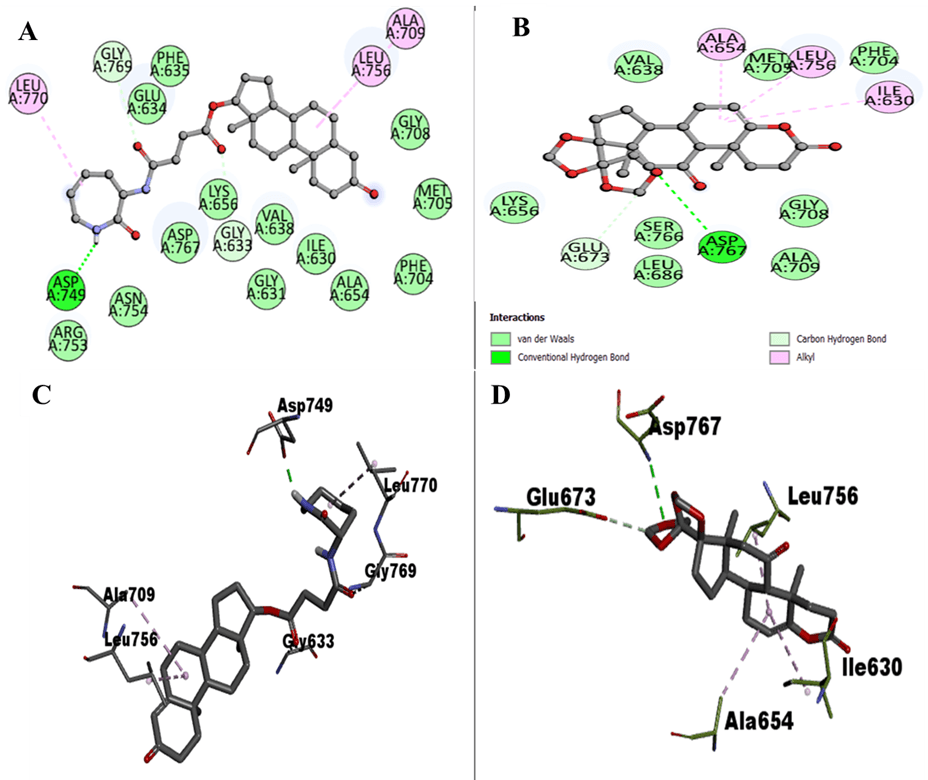
Figure 3. 2D interactions of EphA1 with ZINC12660859 and ZINC12661003, including interacting residue views.
Download figure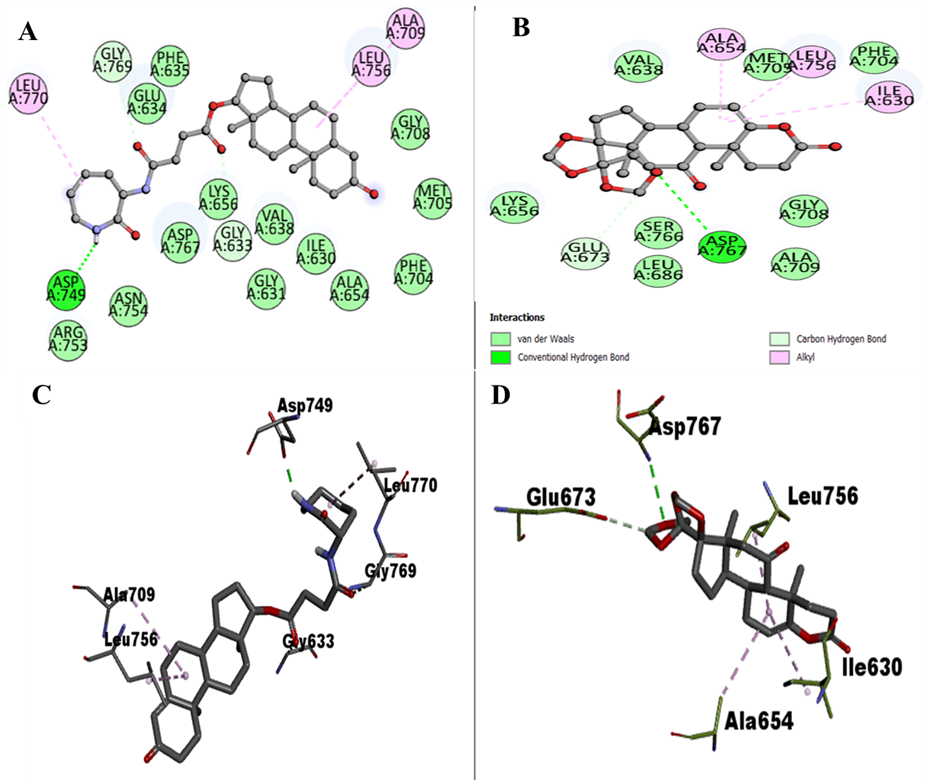{kind=link}
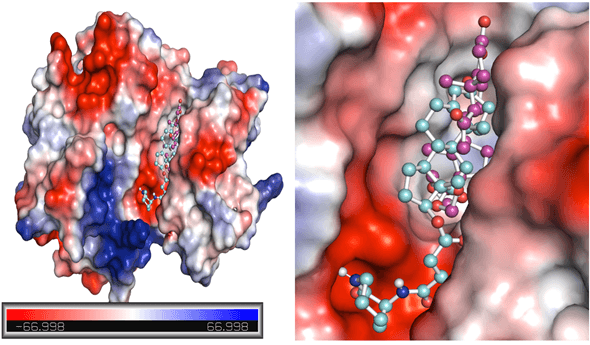
Figure 4. Potential surface representation of EphA1 complexes with ZINC12660859 (cyan) and ZINC12661003 (magenta).
Download figure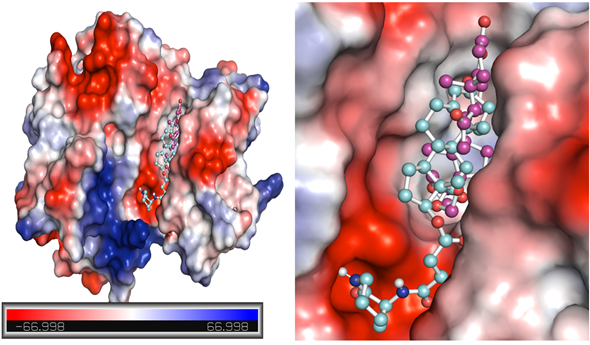{kind=link}
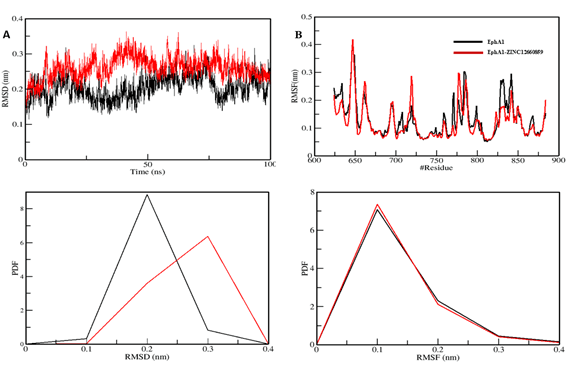
Figure 5. Structural dynamics of the EphA1–ZINC12660859 complex showing RMSD, RMSF, and PDF panels.
Download figure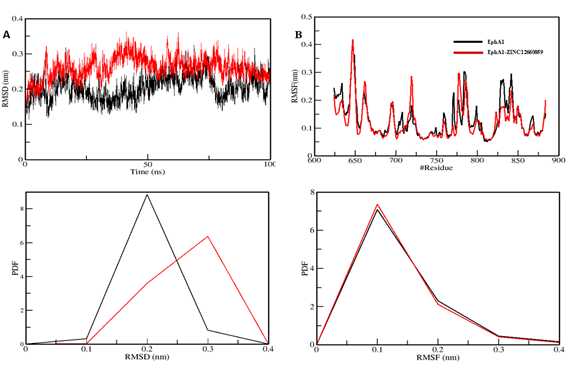{kind=link}
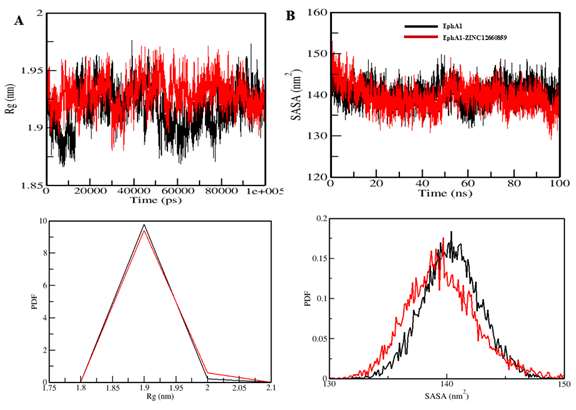
Figure 6. Radius of gyration and solvent-accessible surface area plots for EphA1 upon interaction with ZINC12660859.
Download figure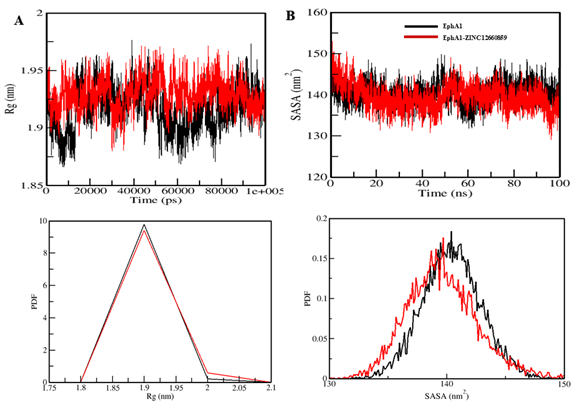{kind=link}
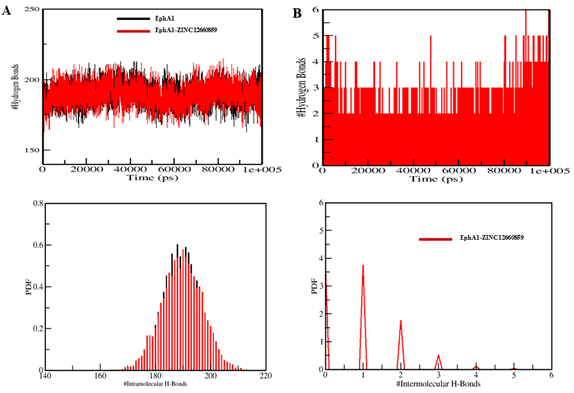
Figure 7. Intramolecular and intermolecular hydrogen-bond analysis for the EphA1–ZINC12660859 system.
Download figure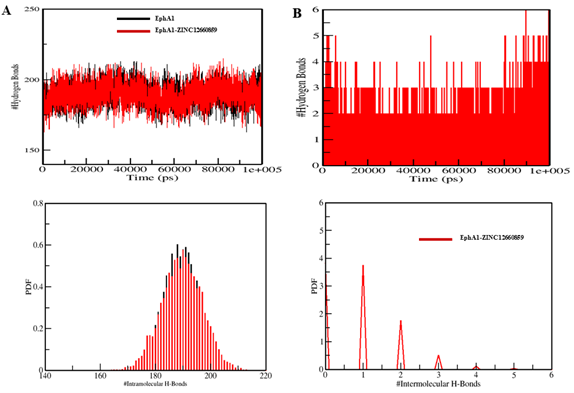{kind=link}
References
- 1
Adu-Gyamfi, E., Czika, A., Liu, T., et al.
Biology of Reproduction article cited in the paper.
Biology of Reproduction · 2021 - 2
Biovia, D.
Discovery Studio Visualizer.
Software reference · 2017 - 3
Dai, B., & Zhang, X.
Neoplasma article cited in the paper.
Neoplasma · 2020 - 4
Daina, A., Michielin, O., & Zoete, V.
Scientific Reports article cited in the paper.
Scientific Reports · 2017 - 5
David, A., Islam, S., Tankhilevich, E., & Sternberg, M.
Journal of Molecular Biology article cited in the paper.
Journal of Molecular Biology · 2022 - 6
DeLano, W.
CCP4 Newsletter on Protein Crystallography article cited in the paper.
CCP4 Newsletter on Protein Crystallography · 2002 - 7
Gajdzis, M., Theocharis, S., Gajdzis, P., et al.
Life article cited in the paper.
Life · 2020 - 8
Hjorthaug, H., & Aasheim, H.
European Journal of Immunology article cited in the paper.
European Journal of Immunology · 2007 - 9
Ieguchi, K., & Maru, Y.
Cancer Science article cited in the paper.
Cancer Science · 2019 - 10
Irwin, J., & Shoichet, B.
Journal of Chemical Information and Modeling article cited in the paper.
Journal of Chemical Information and Modeling · 2005 - 11
Irwin, J., Tang, K., Young, J., et al.
Journal of Chemical Information and Modeling article cited in the paper.
Journal of Chemical Information and Modeling · 2020 - 12
Kang, M., Jeong, W., Bae, H., et al.
Journal of Cellular Physiology article cited in the paper.
Journal of Cellular Physiology · 2018 - 13
Lagunin, A., Stepanchikova, A., Filimonov, D., & Poroikov, V.
Bioinformatics article cited in the paper.
Bioinformatics · 2000 - 14
Li, Y., Yan, H., Wang, F., et al.
Biochemical and Biophysical Research Communications article cited in the paper.
Biochemical and Biophysical Research Communications · 2017 - 15
Liang, Z., Wang, X., Dong, K., et al.
BioMed Research International article cited in the paper.
BioMed Research International · 2021 - 16
Lisabeth, E., Falivelli, G., & Pasquale, E.
Cold Spring Harbor Perspectives in Biology article cited in the paper.
Cold Spring Harbor Perspectives in Biology · 2013 - 17
Ma, J., Wang, Z., Chen, S., et al.
Molecular Neurobiology article cited in the paper.
Molecular Neurobiology · 2021 - 18
Mohammad, T., Mathur, Y., & Hassan, M.
Briefings in Bioinformatics article cited in the paper.
Briefings in Bioinformatics · 2021 - 19
Peng, L., Wang, H., Dong, Y., et al.
International Journal of Clinical and Experimental Pathology article cited in the paper.
International Journal of Clinical and Experimental Pathology · 2013 - 20
Pires, D., Blundell, T., & Ascher, D.
Journal of Medicinal Chemistry article cited in the paper.
Journal of Medicinal Chemistry · 2015 - 21
Rose, P., Prlić, A., Altunkaya, A., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2017 - 22
Toma, M., Erdmann, K., Diezel, M., et al.
PLoS One article cited in the paper.
PLoS One · 2014 - 23
UniProt Consortium
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2019 - 24
Villegas-Llerena, C., Phillips, A., Garcia-Reitboeck, P., Hardy, J., & Pocock, J.
Current Opinion in Neurobiology article cited in the paper.
Current Opinion in Neurobiology · 2016 - 25
Wang, J., Ma, J., Dong, Y., et al.
APMIS article cited in the paper.
APMIS · 2013 - 26
Wang, Y., Dai, Y., Xu, G., et al.
Medical Science Monitor article cited in the paper.
Medical Science Monitor · 2020 - 27
Wang, Y., Yu, H., Shan, Y., et al.
Journal of Experimental & Clinical Cancer Research article cited in the paper.
Journal of Experimental & Clinical Cancer Research · 2016 - 28
Wu, B., Jiang, W., Zhou, D., & Cui, Y.
Anticancer Research article cited in the paper.
Anticancer Research · 2016 - 29
Wu, Y., Du, Z., Mou, J., et al.
Current Medicinal Chemistry article cited in the paper.
Current Medicinal Chemistry · 2022 - 30
Yamazaki, T., Masuda, J., Omori, T., et al.
Journal of Cell Science article cited in the paper.
Journal of Cell Science · 2009 - 31
Yu, L., Ke, J., Du, X., Yu, Z., & Gao, D.
Scientific Reports article cited in the paper.
Scientific Reports · 2019 - 32
Yu, W., & MacKerell, A.D., J.
Methods in Molecular Biology article cited in the paper.
Methods in Molecular Biology · 2017 - 33
Zhang, L., Ai, H., Chen, W., et al.
Scientific Reports article cited in the paper.
Scientific Reports · 2017
Tables
Table 1. List of selected top 66 compounds based on binding affinity towards EphA1.
| S. No. | Ligand ZINC ID | Binding Free Energy (kcal/mol) |
|---|---|---|
| 1 | ZINC03845566 | -11.2 |
| 2 | ZINC08790757 | -11.1 |
| 3 | ZINC08791024 | -11.1 |
| 4 | ZINC08791220 | -11.1 |
| 5 | ZINC08791221 | -11.1 |
| 6 | ZINC12878012 | -11.0 |
| 7 | ZINC12902062 | -11.0 |
| 8 | ZINC12877669 | -10.9 |
| 9 | ZINC12902057 | -10.9 |
| 10 | ZINC12876960 | -10.8 |
| 11 | ZINC12881190 | -10.8 |
| 12 | ZINC08789288 | -10.8 |
| 13 | ZINC08790429 | -10.8 |
| 14 | ZINC08876663 | -10.8 |
| 15 | ZINC12883374 | -10.8 |
| 16 | ZINC08918144 | -10.8 |
| 17 | ZINC08791168 | -10.8 |
| 18 | ZINC08790034 | -10.7 |
| 19 | ZINC08789997 | -10.7 |
| 20 | ZINC08790428 | -10.7 |
| 21 | ZINC09033817 | -10.6 |
| 22 | ZINC08790759 | -10.6 |
| 23 | ZINC08790760 | -10.6 |
| 24 | ZINC08876661 | -10.6 |
| 25 | ZINC12879301 | -10.6 |
| 26 | ZINC02133362 | -10.6 |
| 27 | ZINC08876556 | -10.6 |
| 28 | ZINC11851754 | -10.6 |
| 29 | ZINC12662242 | -10.6 |
| 30 | ZINC12902051 | -10.6 |
| 31 | ZINC08791169 | -10.6 |
| 32 | ZINC03850412 | -10.5 |
| 33 | ZINC08876707 | -10.5 |
| 34 | ZINC04017374 | -10.5 |
| 35 | ZINC08789996 | -10.5 |
| 36 | ZINC08876667 | -10.5 |
| 37 | ZINC12873131 | -10.5 |
| 38 | ZINC12874710 | -10.5 |
| 39 | ZINC12886094 | -10.5 |
| 40 | ZINC12887931 | -10.5 |
| 41 | ZINC12902067 | -10.5 |
| 42 | ZINC08918297 | -10.4 |
| 43 | ZINC02161110 | -10.4 |
| 44 | ZINC04258868 | -10.4 |
| 45 | ZINC08300419 | -10.4 |
| 46 | ZINC08789048 | -10.4 |
| 47 | ZINC12885214 | -10.4 |
| 48 | ZINC03841970 | -10.4 |
| 49 | ZINC08789855 | -10.4 |
| 50 | ZINC12887928 | -10.4 |
| 51 | ZINC08792436 | -10.4 |
| 52 | ZINC08790023 | -10.4 |
| 53 | ZINC03851871 | -10.3 |
| 54 | ZINC08918422 | -10.3 |
| 55 | ZINC08918466 | -10.3 |
| 56 | ZINC12662629 | -10.3 |
| 57 | ZINC03844856 | -10.3 |
| 58 | ZINC12895934 | -10.3 |
| 59 | ZINC04045796 | -10.2 |
| 60 | ZINC04073739 | -10.2 |
| 61 | ZINC12874870 | -10.2 |
| 62 | ZINC00348369 | -10.2 |
| 63 | ZINC02090582 | -10.2 |
| 64 | ZINC04237100 | -10.2 |
| 65 | ZINC12660859 | -9.9 |
| 66 | ZINC12661003 | -9.7 |
Table 2. The predicted druglike properties of four potentially selective inhibitors of EphA1.
| Compound ID | Molecular Weight | Rotatable Bonds | H-Bond Donor | H-Bond Acceptor | LogP | PAINS |
|---|---|---|---|---|---|---|
| ZINC12660859 | 498.664 | 5 | 2 | 5 | 3.9952 | 0 |
| ZINC12661003 | 404.459 | 0 | 0 | 7 | 2.6825 | 0 |
Table 3. Predicted ADMET characteristics of chosen hits.
| ZINC ID | GI Absorption | BBB Permeation | CYP2D6 Inhibitor | OCT2 Substrate | AMES Toxicity |
|---|---|---|---|---|---|
| ZINC12660859 | 94.952 (High) | -0.177 | No | No | No |
| ZINC12661003 | 99.476 (High) | -0.653 | No | No | No |
Table 4. PASS analysis results: Activities with Pa and Pi values of 2 selected compounds.
| S. No. | Compound | Pa value | Pi value | Biological Activity |
|---|---|---|---|---|
| 1 | ZINC12660859 | 0,845 | 0,025 | CYP2C12 substrate |
| 1 | ZINC12660859 | 0,500 | 0,027 | AR expression inhibitor |
| 1 | ZINC12660859 | 0,464 | 0,083 | Antineoplastic |
| 2 | ZINC12661003 | 0,566 | 0,053 | Antineoplastic |
| 2 | ZINC12661003 | 0,649 | 0,027 | Phosphatase inhibitor |
| 2 | ZINC12661003 | 0,488 | 0,016 | Myc inhibitor |
| 2 | ZINC12661003 | 0,388 | 0,079 | Apoptosis agonist |
| 2 | ZINC12661003 | 0,339 | 0,066 | Antimetastatic |
Table 5. Interacting residues analyzed using Discovery Studio.
| S. No. | Compound ID | H BONDING | Other Interactions |
|---|---|---|---|
| 1 | ZINC12660859 | Asp749, Gly769, Gly633 | Lys656, Arg753, Asn754, Asp767, Val638, Ile630, Leu770, Leu756, Ala709, Gly631, Ala654, Phe704, Met705, Gly708, Phe635, Glu634 |
| 2 | ZINC12661003 | Asp767, Glu673 | Lys656, Ser766, Leu686, Ala709, Gly708, Val638, Met705, Phe704, Ala654, Leu756, Ile630 |
Table 6. The mean values of various MD parameters were computed based on 100 ns simulations.
| System | RMSD (nm) | RMSF (nm) | Rg (nm) | SASA (nm2) | # H-Bonds |
|---|---|---|---|---|---|
| EphA1 | 0.20 | 0.13 | 1.92 | 140.42 | 189 |
| EphA1-ZINC12660859 | 0.26 | 0.12 | 1.93 | 139.62 | 190 |