


About Article
Impact of Single Nucleotide Polymorphisms in TERF1-Interacting Nuclear Factor 2 and Their Association with Dyskeratosis Congenita Autosomal Dominant-3
Volume 1, No. 1 · 2026
Highlights
- This study analyzed 271 non-synonymous TINF2 variants to identify mutations potentially associated with autosomal dominant dyskeratosis congenita.
- Sequence-based screening used SIFT, PROVEAN, PolyPhen-2, Mutation Assessor, and SAAFEC-SEQ, while structure-based stability analysis used SDM2, DUET, mCSM, and MUpro.
- Of 134 structurally mappable variants in PDB 5XYF, 35 high-confidence deleterious and destabilizing mutations were retained after consensus filtering.
- Twelve missense variants—A16D, V22G, V34G, L38P, V49G, R52P, R56G, E109G, F114S, L121P, Y139C, and L150P—were consistently predicted as pathogenic.
- Structural analyses indicated altered packing density, disrupted noncovalent interactions, and aggregation or solubility changes that may destabilize shelterin complex function.
Abstract
Dyskeratosis congenita (DC) is a rare hereditary telomere biology disorder characterized by bone marrow failure, nail dystrophy, mucosal leukoplakia, and abnormal skin pigmentation.
The study focused on TERF1-interacting nuclear factor 2 (TIN2), encoded by TINF2, a key shelterin-complex component that coordinates TRF1, TRF2, and TPP1/POT1 interactions and whose heterozygous missense mutations cause autosomal dominant dyskeratosis congenita type 3.
A total of 271 non-synonymous SNPs were evaluated with sequence-based tools, and 134 variants within the resolved crystal structure (PDB ID: 5XYF) were further analyzed with structure-based stability predictors.
High-confidence deleterious variants were assessed with PMut, PhD-SNP, and Rhapsody, yielding 12 consistently pathogenic missense mutations.
The selected mutations showed altered stability, packing density, surface accessibility, aggregation propensity, and interatomic interactions, suggesting disruption of shelterin stability and telomere maintenance.
Keywords
Article Overview
This article examines how missense variation in TINF2 may disrupt TIN2, a central scaffolding component of the shelterin complex that protects telomeres and regulates telomerase-dependent telomere length.
The paper frames dyskeratosis congenita as a telomere biology disorder and argues that even modest amino-acid substitutions in TIN2 can weaken protein stability, alter intermolecular binding, and compromise chromosome-end protection.
To prioritize the most disease-relevant variants, the study integrates multiple sequence-based, structure-based, and pathogenicity-prediction tools, followed by structural characterization of stability, packing, solvent accessibility, interatomic interactions, and aggregation propensity.
1. About the Article
This is a research article published in Clinical & Molecular Biomedicine that reports a comprehensive in silico analysis of TINF2 missense variation and its relevance to autosomal dominant dyskeratosis congenita type 3.
The work is authored by researchers from Yeungnam University and Jamia Millia Islamia, with Sarfraz Ahmed and Kim Ji Hoe listed as corresponding authors.
- Journal: Clinical & Molecular Biomedicine
- Volume/Issue: Vol. 1, No. 1
- Header year: 2026
- Authors: Sarfraz Ahmed; Lalita Thangwal; Kim Ji Hoe
- Affiliation 1: Department of Medical Biotechnology and Research Institute of Cell Culture, Yeungnam University, Gyeongsan 38541, Republic of Korea
- Affiliation 2: Department of Computer Science, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India
- Corresponding authors: Sarfraz Ahmed; Kim Ji Hoe
- Corresponding emails: sarfrazahmed6954@yu.ac.kr; kimjihoe@ynu.ac.kr
- Received: April 16th, 2024
- Accepted: March 31, 2026
- Published: May 21, 2024
- License: Creative Commons CC-BY 4.0
- Open-access status: Open Access
- Header page range on first page: pp. 1–25
- Visible article page range in the PDF: 1–6
2. Introduction
The introduction explains that telomeres are protected by the six-subunit shelterin complex and identifies TIN2 as the central bridging component linking TRF1, TRF2, and TPP1/POT1.
It emphasizes that TIN2 consists of 354 amino acids and is essential for telomere-length regulation, TRF1 stabilization, and chromosome-end protection.
The article connects TINF2 mutation to dyskeratosis congenita, particularly autosomal dominant type 3, and notes prior hotspot regions around residues 269–298.
The rationale for the study is that computational integration across multiple predictive tools can prioritize variants likely to disturb TIN2 structure, shelterin assembly, and telomere maintenance.
- Target gene: TINF2
- Target protein: TIN2
- Protein length noted: 354 amino acids
- Complex discussed: shelterin
- Disease focus: dyskeratosis congenita autosomal dominant-3
- Structure discussed: TRFH-like N-terminal region and TRF1-binding-related regions
3. Materials and Methods
TINF2 sequence data were retrieved from UniProt (Q9BSI4), while missense variants were aggregated from dbSNP, HGMD, ClinVar, and Ensembl. The resolved crystal structure used for structural analysis was PDB ID 5XYF.
All 271 non-synonymous variants were screened with five sequence-based predictors: SIFT, PolyPhen-2, PROVEAN, Mutation Assessor, and SAAFEC-SEQ.
Only the 134 variants that map to the resolved N-terminal structure were further evaluated with four structure-based tools: SDM2, DUET, mCSM, and MUpro.
Potential disease association was then tested with PMut, PhD-SNP, and Rhapsody, and follow-up analyses included SDM2-based packing and surface metrics, Arpeggio-based interatomic interaction analysis, and SODA-based aggregation/solubility analysis.
- UniProt ID: Q9BSI4
- Protein structure: PDB ID 5XYF
- Approximate total mutations collected from Ensembl: 900
- Non-synonymous mutations analyzed: 271
- Structure-mappable nsSNPs analyzed: 134
- Sequence-based tools: SIFT; PolyPhen-2; PROVEAN; Mutation Assessor; SAAFEC-SEQ
- Structure-based tools: SDM2; DUET; mCSM; MUpro
- Pathogenicity tools: PMut; PhD-SNP; Rhapsody
- Interaction tool: Arpeggio
- Aggregation tool: SODA
2.1. Data Retrieval
The study assembled TINF2 missense variants from multiple genomic and clinical databases and removed duplicates before prioritization.
Only the resolved N-terminal segment present in PDB 5XYF was eligible for downstream structure-based analysis.
2.2. Prediction of deleterious mutations using sequence-based tools
Five predictors were used to assess functional and free-energy consequences of amino-acid substitutions using evolutionary conservation and sequence-derived physicochemical features.
Thresholds were explicitly described for SIFT, PROVEAN, and Mutation Assessor, while PolyPhen-2 and SAAFEC-SEQ were used as complementary evidence sources.
2.3. Prediction of the destabilizing mutation using structure-based tools
SDM2, mCSM, DUET, and MUpro were used to estimate stability changes, chiefly through predicted ΔΔG shifts or confidence scores reflecting increases or decreases in stability.
These tools focused on the subset of variants that fall within the resolved crystal structure.
2.4. Identification of Pathogenic nsSNPs
PMut, PhD-SNP, and Rhapsody were used to distinguish disease-associated variants from neutral substitutions using structure- and sequence-aware models.
2.5. Analysis of packing density and accessible surface area by using SDM2
The updated SDM2 workflow was also used to examine relative solvent accessibility, occluded surface packing, and residue depth for wild-type and mutant states.
2.6. Analysis of noncovalent interactions
Arpeggio was used to quantify hydrogen-bonding, hydrophobic, ionic, aromatic, and van der Waals contacts to compare mutant and wild-type local interaction networks.
2.7. Analysis of aggregation propensity
SODA was used to estimate mutation-associated changes in solubility and aggregation propensity using sequence or structure as input.
4. Results and Discussion
The article reports 271 TINF2 nsSNPs after filtering the broader mutation list, with 134 variants located in the resolved N-terminal structure used for structure-based analysis.
Consensus sequence-based analysis showed substantial tool-to-tool variation, but consensus filtering prioritized variants predicted as deleterious by at least four of five methods.
Consensus structure-based filtering further narrowed the set to 35 high-confidence deleterious and destabilizing variants, of which 12 were consistently predicted as pathogenic across disease-association tools.
The selected mutations were then characterized for packing-density changes, altered interatomic contacts, and aggregation or solubility shifts, supporting a model in which TIN2 destabilization can impair shelterin integrity and telomere protection.
- Total Ensembl mutations noted: about 900
- Non-synonymous mutations analyzed: 271
- Structure-mappable variants: 134
- High-confidence deleterious and destabilizing variants after stringent filtering: 35
- Final pathogenic variants prioritized: 12
3.1. Identification of Non-Synonymous Variants in TINF2
The paper establishes the full screening set, explains why only residues within the resolved 5XYF structure could undergo structure-based analysis, and describes the multi-level prioritization workflow.
3.2. Sequence-Based Prediction of Deleterious Mutations
SIFT predicted 142 variants as damaging, PolyPhen-2 identified 96 as possibly or probably damaging, PROVEAN identified 41 as deleterious, Mutation Assessor identified 80 as functionally impactful, and SAAFEC-SEQ predicted 264 variants to reduce folding free energy.
Variants predicted as deleterious by at least four of the five tools were classified as high-confidence functionally damaging substitutions.
- SIFT deleterious: 142
- PolyPhen-2 damaging: 96
- PROVEAN deleterious: 41
- Mutation Assessor impactful: 80
- SAAFEC-SEQ reduced folding free energy: 264
3.3. Structure-Based Stability Analysis
Among 134 structure-mappable variants, SDM2 predicted 58 destabilizing substitutions, DUET identified 103, mCSM identified 120, and MUpro predicted 128 to decrease stability.
Only variants destabilizing in at least three of the four tools and deleterious in at least four of the five sequence-based tools were retained, yielding 35 high-confidence variants.
- SDM2 destabilizing: 58
- DUET destabilizing: 103
- mCSM destabilizing: 120
- MUpro decreasing stability: 128
- Final high-confidence deleterious and destabilizing variants: 35
3.4. Identification of pathogenic nsSNPs
PMut classified 25 of the 35 high-confidence variants as pathogenic, PhD-SNP classified 24, and Rhapsody classified 11 as disease-associated.
Twelve variants—A16D, V22G, V34G, L38P, V49G, R52P, R56G, E109G, F114S, L121P, Y139C, and L150P—were consistently selected for detailed structural analysis.
- PMut pathogenic: 25
- PhD-SNP pathogenic: 24
- Rhapsody disease-associated: 11
- Prioritized variants: A16D; V22G; V34G; L38P; V49G; R52P; R56G; E109G; F114S; L121P; Y139C; L150P
3.4.1. Changes in Stability and Packing Density
The analysis of RSA, residue depth, and OSP suggested that many selected variants alter local packing, especially when hydrophobic residues are replaced by glycine or proline.
The paper particularly notes that proline substitutions such as L38P, L121P, and L150P are likely disruptive because of conformational rigidity and helix-breaking tendencies.
3.4.2. Alterations in Noncovalent Interactions
Arpeggio-based analysis showed that several mutations reduce hydrogen-bonding, hydrophobic, and aromatic contacts relative to wild type, which may destabilize local structure or protein–protein interfaces.
Charged-residue substitutions such as R52P, R56G, and E109G were highlighted as potentially disruptive to electrostatic interactions relevant to TRF1 and TRF2 binding.
3.4.3. Aggregation Propensity and Solubility Changes
SODA analysis indicated that nine of the twelve prioritized mutations were more soluble while three were less soluble, but the text interprets the overall profile as increased aggregation tendency or solubility disruption capable of impairing shelterin assembly.
The three variants explicitly labeled less soluble in Table 3 are A16D, Y139C, and L150P.
- Less-soluble variants in Table 3: A16D; Y139C; L150P
5. Conclusion
The paper concludes that a systematic in silico framework can prioritize TINF2 variants likely to destabilize TIN2 and contribute to dyskeratosis congenita pathogenesis.
The twelve prioritized missense variants are proposed to alter packing density, noncovalent interactions, and aggregation-related behavior in ways that could weaken shelterin assembly and telomere protection.
The authors emphasize that the computational findings are mechanistically informative but still require experimental validation.
- Main outcome: prioritization of 12 pathogenic TINF2 missense variants
- Biological implication: impaired shelterin stability and telomere protection
- Next step: experimental validation
6. Statements and Declarations
The article includes standard closing sections for conflict of interest, data availability, funding, acknowledgements, supplementary materials, and author contributions.
- Conflict of Interest: There is no conflict of interest to declare.
- Data Availability Statement: The data supporting this study are provided in this article.
- Funding: None
- Acknowledgements: None
- Supplementary Materials: None
Figures
The PDF contains four figures showing the computational workflow, mutation distribution, and graphical summaries of sequence-based and structure-based prediction results.
Figure 1

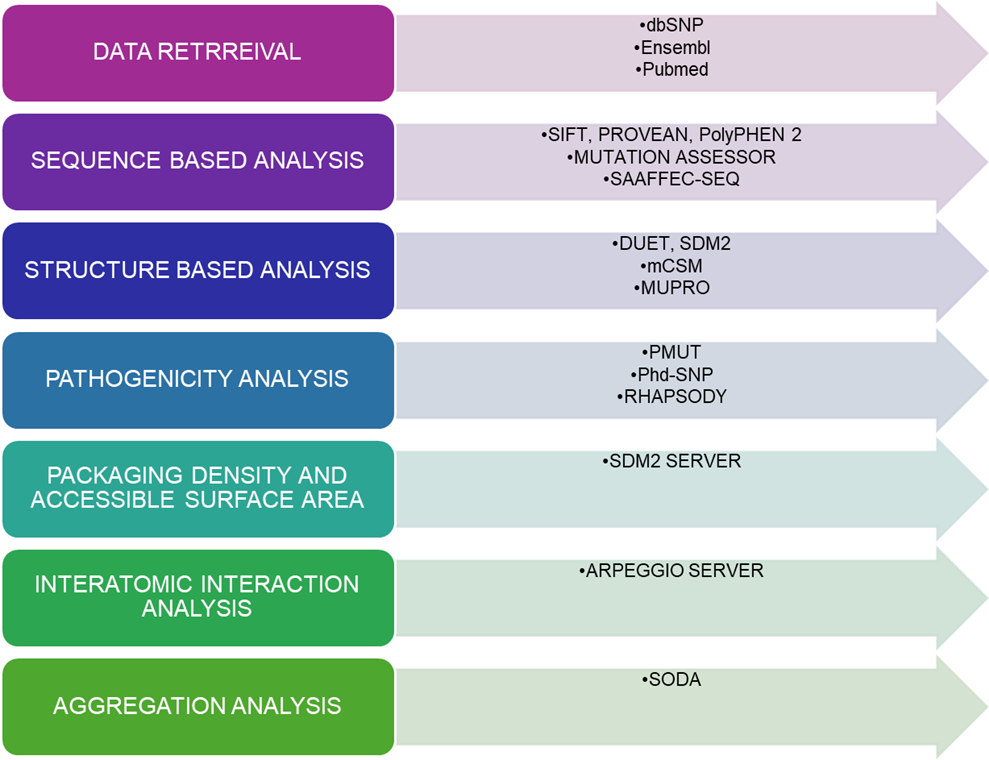{kind=link}
Figure 1. Overview of the computational aspects to predict the pathogenic mutation in TINF2.
Download figure
Figure 2. Representation of the number of SNPs in TINF2 using the Ensembl database.
Download figure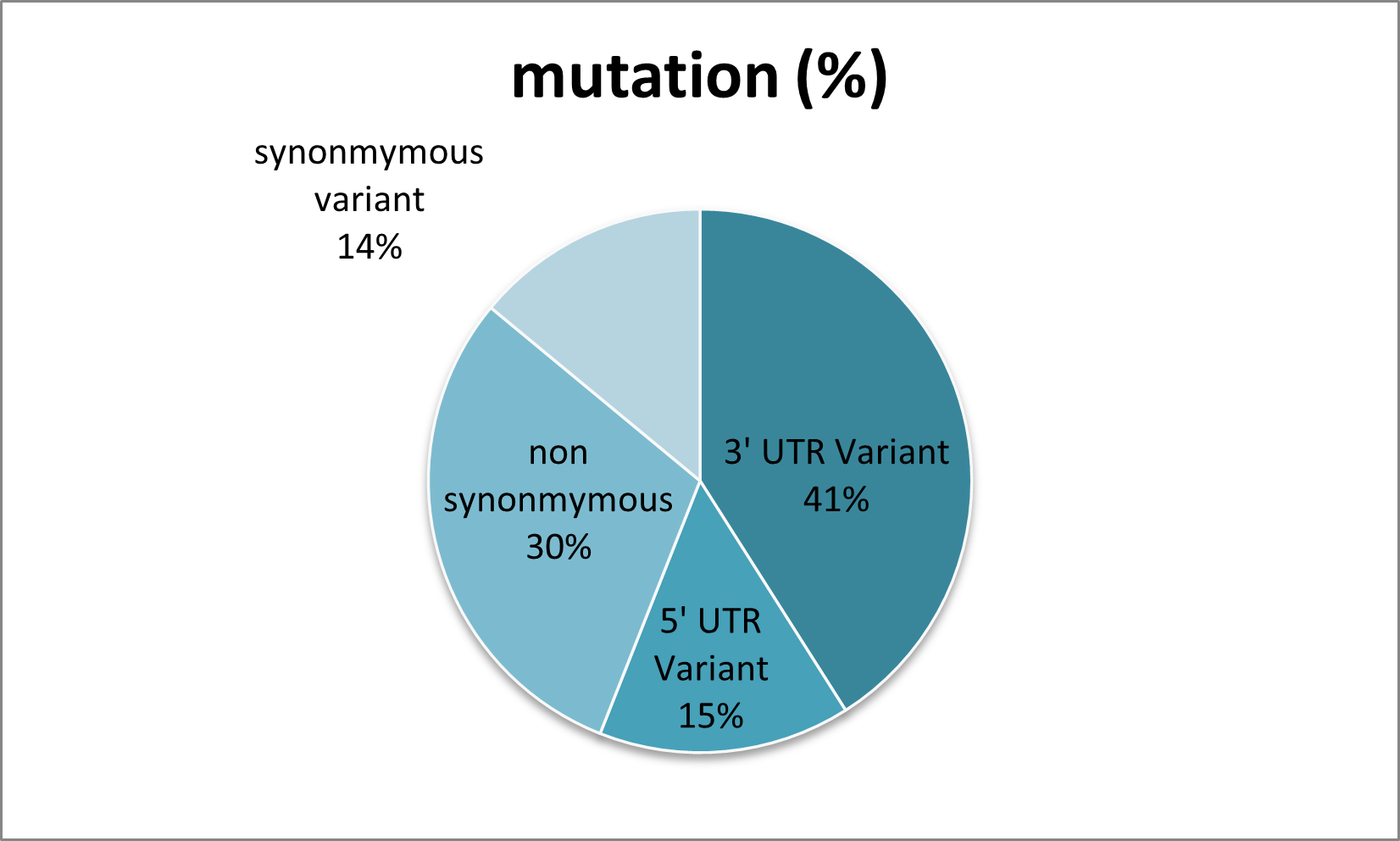{kind=link}

Figure 3. Graphical representation of deleterious nsSNPs predicted by sequence-based tools.
Download figure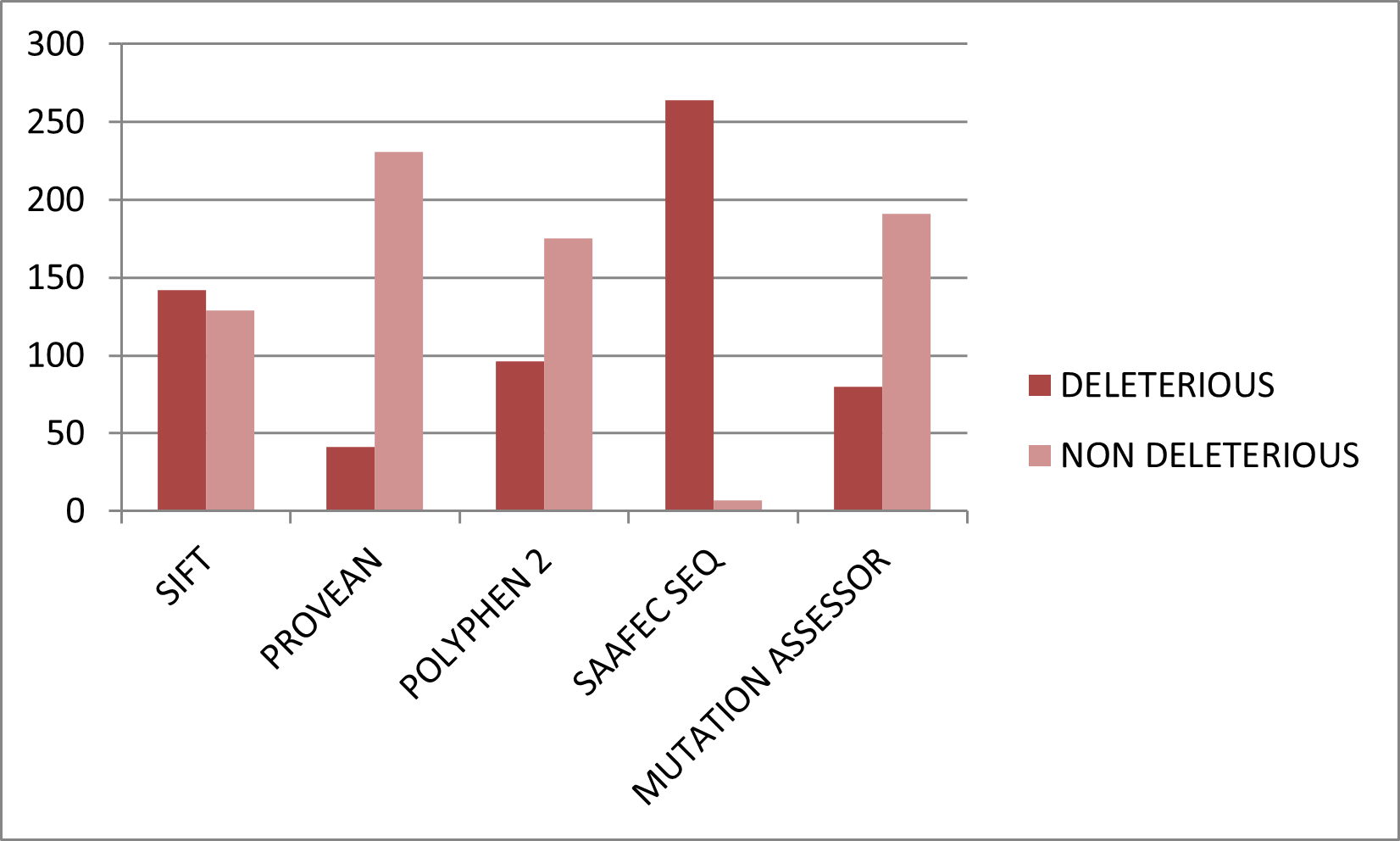{kind=link}

Figure 4. Graphical representation of deleterious and neutral nsSNPs predicted by structure-based tools.
Download figure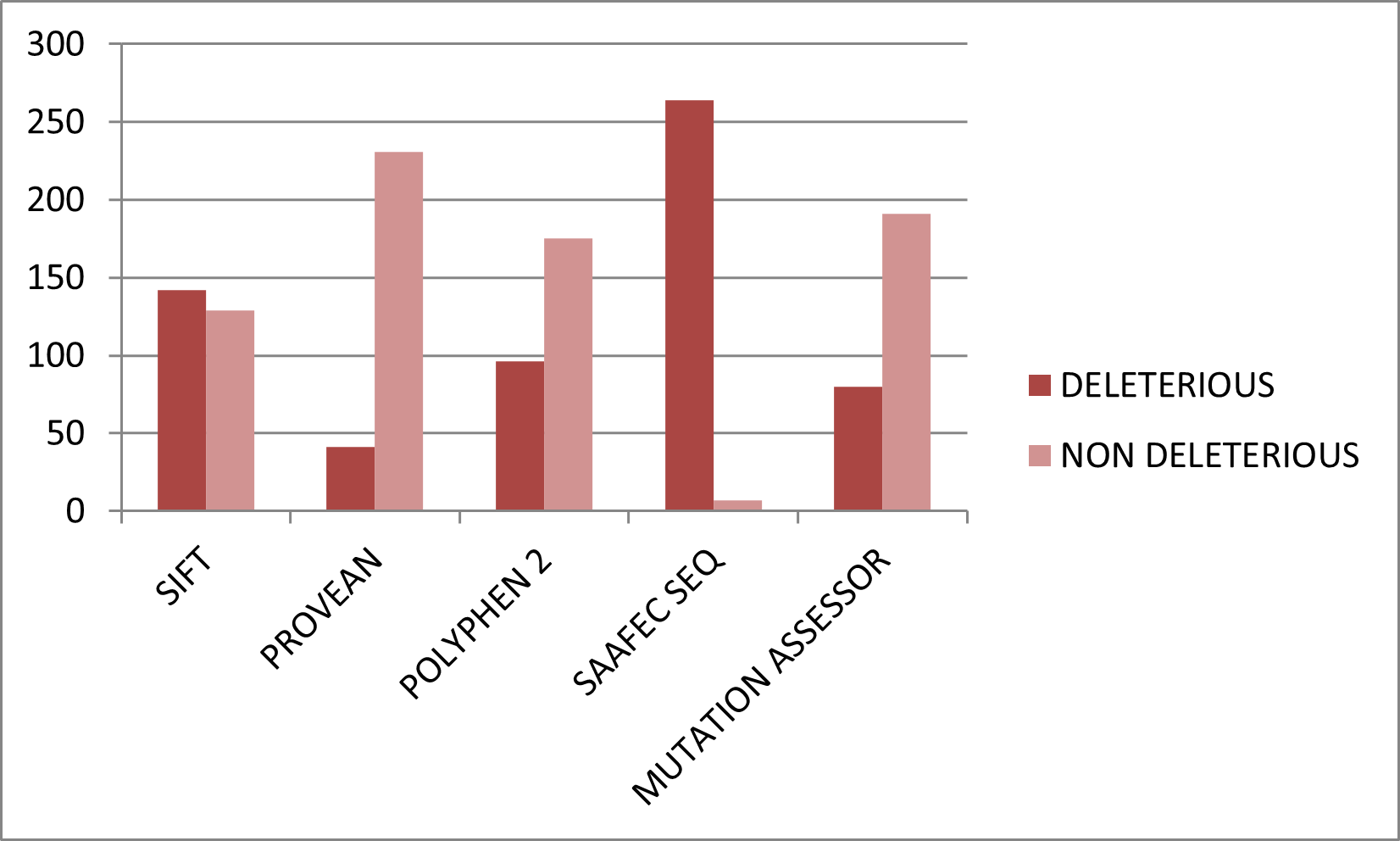{kind=link}
References
- 1
Berman, H. M., Battistuz, T., Bhat, T. N., et al.
Acta Crystallographica Section D article cited in the paper.
Acta Crystallographica Section D · 2002 - 2
Cheng, J., Randall, A., & Baldi, P.
Proteins article cited in the paper.
Proteins · 2006 - 3
Choi, Y., & Chan, A. P.
Bioinformatics article cited in the paper.
Bioinformatics · 2015 - 4
de Lange, T.
Genes & Development article cited in the paper.
Genes & Development · 2005 - 5
DeDecker, B. S., O’Brien, R., Fleming, P. J., et al.
Journal of Molecular Biology article cited in the paper.
Journal of Molecular Biology · 1996 - 6
Frescas, D., & de Lange, T.
Journal of Biological Chemistry article cited in the paper.
Journal of Biological Chemistry · 2014 - 7
Getov, I., Petukh, M., & Alexov, E.
Bioinformatics article cited in the paper.
Bioinformatics · 2016 - 8
Hubbard, T., Barker, D., Birney, E., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2002 - 9
Jubb, H. C., Ochoa-Montaño, B., Pitt, W. R., Ascher, D. B., & Blundell, T. L.
Journal of Molecular Biology article cited in the paper.
Journal of Molecular Biology · 2017 - 10
Kumar, P., Henikoff, S., & Ng, P. C.
Nature Protocols article cited in the paper.
Nature Protocols · 2009 - 11
Landrum, M. J., Lee, J. M., Riley, G. R., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2014 - 12
López-Ferrando, V., Gazzo, A., de la Cruz, X., Orozco, M., & Gelpí, J. L.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2017 - 13
Paladin, L., Piovesan, D., & Tosatto, S. C. E.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2017 - 14
Pandurangan, A. P., Ochoa-Montaño, B., Ascher, D. B., & Blundell, T. L.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2017 - 15
Pike, A. M., Strong, M. A., Ouyang, J. P. T., & Greider, C. W.
Molecular Cell article cited in the paper.
Molecular Cell · 2019 - 16
Pires, D. E. V., Ascher, D. B., & Blundell, T. L.
Bioinformatics and Nucleic Acids Research articles cited in the paper.
Bioinformatics / Nucleic Acids Research · 2013–2014 - 17
Ponzoni, L., Peñaherrera, D. A., Oltvai, Z. N., & Bahar, I.
Bioinformatics article cited in the paper.
Bioinformatics · 2020 - 18
Ramensky, V., Bork, P., & Sunyaev, S.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2002 - 19
Reva, B., Antipin, Y., & Sander, C.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2011 - 20
Richards, F. M., & Lim, W. A.
Quarterly Reviews of Biophysics article cited in the paper.
Quarterly Reviews of Biophysics · 1993 - 21
Savage, S. A., Giri, N., Baerlocher, G. M., et al.
Nature Genetics article cited in the paper.
Nature Genetics · 2008 - 22
Sherry, S. T., Ward, M. H., Kholodov, M., et al.
Nucleic Acids Research article cited in the paper.
Nucleic Acids Research · 2001 - 23
Stenson, P. D., Mort, M., Ball, E. V., et al.
Genome Medicine article cited in the paper.
Genome Medicine · 2009 - 24
Yang, D., He, Q., Kim, H., Ma, W., & Songyang, Z.
Journal of Biological Chemistry article cited in the paper.
Journal of Biological Chemistry · 2011
Tables
Table 1. Prediction of RSA, residue depth, and OSP for the wild-type and mutant TINF2 proteins using the SDM2 server.
| S. No. | Mutation | WT RSA (%) | WT Depth (Å) | WT OSP | MT RSA (%) | MT Depth (Å) | MT OSP | Outcome |
|---|---|---|---|---|---|---|---|---|
| 1 | A16D | 0.0 | 8.1 | 0.60 | 0.0 | 8.2 | 0.65 | Increased Solubility |
| 2 | V22G | 9.9 | 4.6 | 0.54 | 13.6 | 5.2 | 0.44 | Reduced Solubility |
| 3 | V34G | 0.4 | 7.5 | 0.53 | 17.0 | 7.0 | 0.47 | Increased Solubility |
| 4 | L38P | 0.3 | 8.6 | 0.52 | 6.2 | 8.1 | 0.51 | Increased Solubility |
| 5 | V49G | 7.1 | 5.1 | 0.42 | 34.4 | 4.1 | 0.26 | Increased Solubility |
| 6 | R52P | 37.7 | 3.9 | 0.31 | 13.3 | 4.8 | 0.42 | Reduced Solubility |
| 7 | R56G | 44.7 | 3.6 | 0.29 | 70.6 | 4.0 | 0.34 | Increased Solubility |
| 8 | E109G | 85.2 | 3.3 | 0.22 | 94.3 | 3.5 | 0.34 | Increased Solubility |
| 9 | F114S | 7.2 | 6.1 | 0.49 | 21.6 | 5.2 | 0.37 | Increased Solubility |
| 10 | L121P | 5.6 | 5.9 | 0.52 | 13.8 | 5.5 | 0.51 | Increased Solubility |
| 11 | Y139C | 12.9 | 4.8 | 0.49 | 9.1 | 5.3 | 0.45 | Reduced Solubility |
| 12 | L150P | 4.4 | 5.3 | 0.53 | 5.5 | 5.7 | 0.56 | Increased Solubility |
Table 2. Arpeggio server estimates of interatomic interactions for WT and mutant protein structures.
| S. No. | Variants | Vander Waals | Hydrogen | Ionic | Aromatics | Hydrophobic |
|---|---|---|---|---|---|---|
| 1 | Wild Type | 175 | 267 | 17 | 18 | 471 |
| 2 | A16D | 176 | 267 | 17 | 18 | 471 |
| 3 | V22G | 174 | 267 | 17 | 18 | 464 |
| 4 | V34G | 175 | 267 | 17 | 18 | 464 |
| 5 | L38P | 175 | 263 | 17 | 18 | 463 |
| 6 | V49G | 174 | 267 | 17 | 18 | 459 |
| 7 | R52P | 176 | 267 | 17 | 18 | 472 |
| 8 | R56G | 175 | 267 | 17 | 18 | 470 |
| 9 | E109G | 175 | 267 | 17 | 18 | 471 |
| 10 | F114S | 173 | 266 | 17 | 14 | 446 |
| 11 | L121P | 173 | 263 | 17 | 18 | 465 |
| 12 | Y139C | 172 | 268 | 17 | 13 | 451 |
| 13 | L150P | 175 | 268 | 17 | 18 | 472 |
Table 3. Prediction of aggregation propensity of mutant TINF2 protein using the SODA server.
| S. No. | Mutation | SODA | Remark |
|---|---|---|---|
| 1 | A16D | -2.9 | Less Soluble |
| 2 | V22G | 20.6 | More Soluble |
| 3 | V34G | 13.4 | More Soluble |
| 4 | L38P | 5.9 | More Soluble |
| 5 | V49G | 18.9 | More Soluble |
| 6 | R52P | 13.9 | More Soluble |
| 7 | R56G | 7.74 | More Soluble |
| 8 | E109G | 1.59 | More Soluble |
| 9 | F114S | 9.37 | More Soluble |
| 10 | L121P | 1.3 | More Soluble |
| 11 | Y139C | -6.5 | Less Soluble |
| 12 | L150P | -8.1 | Less Soluble |