


About Article
Genome-wide annotation and structural modeling of hypothetical proteins in Listeria monocytogenes
Vol. 1, No. 1 · 2026
Highlights
- The article investigates how missense mutations in the ARSB gene destabilize protein structure and contribute to mucopolysaccharidosis type VI (MPS VI).
- The study combines sequence-based prediction, structure-based stability analysis, pathogenicity scoring, conservation analysis, aggregation propensity analysis, and molecular dynamics simulation.
- The abstract reports 430 nsSNPs screened, 141 variants analyzed structurally, 57 overlapping high-confidence variants, and 44 highly deleterious mutations identified.
- ConSurf analysis highlighted 12 final mutations in highly conserved regions, while SODA and structural interaction analysis emphasized A237D and W353R as key low-solubility, aggregation-relevant variants.
- The PDF contains several internal inconsistencies in dates and variant counts, so this JSON preserves those conflicts explicitly instead of silently normalizing them.
Abstract
Mutations in the arylsulfatase B (ARSB) gene are directly implicated in mucopolysaccharidosis type VI (MPS VI). Several non-synonymous single-nucleotide polymorphisms (nsSNPs) in ARSB have been associated with disease pathogenesis. A comprehensive evaluation of these variants is essential to understand their structural and functional consequences. In this study, a systematic in silico analysis was performed to identify deleterious nsSNPs in the ARSB gene. Initially, 430 nsSNPs were evaluated using sequence-based prediction tools, including SIFT, PolyPhen-2, FATHMM, and Mutation Assessor. Subsequently, 141 nsSNPs were subjected to structure-based stability analysis using MAESTROweb, SDM2, mCSM, and DynaMut2, of which 57 variants overlapped with previous reports. High-confidence deleterious nsSNPs were further assessed for pathogenicity using PMut and MutPred2 servers. Our integrated computational approach identified 44 highly deleterious mutations. Aggregation propensity analysis revealed that 29 of these variants exhibit increased aggregation tendencies, while one variant demonstrated progressive loss of solubility. Molecular dynamics simulations further indicated that high- confidence deleterious nsSNPs significantly disrupt ARSB structural integrity, enhance molecular flexibility, reduce structural rigidity, and promote atomic-level aggregation. Overall, this study provides mechanistic insights into how pathogenic mutations destabilize the ARSB protein and contribute to MPS VI pathogenesis, highlighting potential targets for future therapeutic investigation.
Keywords
Article Overview
The article introduces lysosomal storage diseases as disorders caused by deficiency of specific lysosomal enzymes and places MPS VI within that group as an autosomal recessive disease caused by ARSB mutations.
ARSB encodes N-acetylgalactosamine-4-sulfatase, which removes sulfate groups from dermatan sulfate and chondroitin-4-sulfate. Loss of ARSB function leads to glycosaminoglycan accumulation and progressive tissue pathology affecting the skeleton, joints, cornea, heart, liver, spleen, and respiratory system.
The paper emphasizes that more than 200 ARSB variants have been reported but that the structural and functional effects of many variants remain incompletely characterized, which complicates prognosis and therapeutic planning.
To address that gap, the study applies a computational workflow integrating sequence, structure, conservation, aggregation, and dynamics analyses to prioritize pathogenic ARSB nsSNPs and explain their mechanistic contribution to disease.
1. About the Article
This is a research article published in Cellular & Molecular Intelligence. It studies mutation-induced instability of arylsulfatase B (ARSB) in the context of mucopolysaccharidosis type VI.
The authors are affiliated with Jamia Hamdard, New Delhi, and King Faisal Specialist Hospital and Research, Riyadh. The PDF identifies Mohammad Asim Azhar as corresponding author in the affiliation block, but the email line names Barka Basharat together with that email address.
- Journal: Cellular & Molecular Intelligence
- Volume/Issue shown: Vol. 1, No. 1
- Publication year shown in header: 2026
- Article title: Molecular Insights into Arylsulfatase B Mutation-Induced Instability in Mucopolysaccharidosis Type VI
- Authors: Barka Basharat; Nushrat Jahan; Mohammad Asim Azhar
- Affiliation 1: Department of Biotechnology, School of Chemical and Life Sciences, Jamia Hamdard, Hamdard Nagar, New Delhi, India
- Affiliation 2: Organ Transplant Center of Excellence, King Faisal Specialist Hospital and Research, Riyadh, Kingdom of Saudi Arabia
- Corresponding email shown: azharasim@gmail.com
- Received: April 16th, 2024
- Accepted: March 31, 2026
- Published: May 21, 2024
- License statement: Creative Commons CC-BY 4.0
- Open-access status: Open Access
- Header page range shown on first page: 1000–1025
- Printed article page range visible in pages: 1000–1007
2. Introduction
The introduction explains lysosomal storage diseases as progressive disorders caused by deficiency of lysosomal enzymes or associated proteins, leading to substrate accumulation and multisystem disease. MPS VI is described specifically as Maroteaux-Lamy syndrome caused by ARSB mutations.
ARSB hydrolyzes sulfate groups from dermatan sulfate and chondroitin-4-sulfate. Deficient activity causes glycosaminoglycan buildup and progressive manifestations such as dysostosis multiplex, joint stiffness, corneal clouding, cardiac valve disease, hepatosplenomegaly, and respiratory complications.
Clinical severity varies, with severe cases presenting early in life and progressing to major disability. Diagnosis is described as combining clinical evaluation, urinary GAG quantification, enzymatic assays, and ARSB genotyping.
The authors argue that ARSB nsSNPs can disrupt protein stability, folding, or interactions, and they position the study as a systematic effort to evaluate such variants using a computational prioritization pipeline. Figure 1 on page 2 depicts that multi-step pipeline across sequence-based, structure-based, disease-prediction, and aggregation analyses.
- Human ARSB UniProt ID used in the study: P15848
- Crystal structure used in the study: PDB ID 1FSU
- Figure 1 shows the computational workflow for forecasting pathogenicity in ARSB
- The introduction mentions more than 200 reported ARSB variants
3. Materials and Methods
The methods section describes a fully computational analysis workflow built around mutation retrieval, sequence-based deleteriousness prediction, structure-based stability estimation, pathogenicity scoring, conservation analysis, aggregation propensity analysis, interaction analysis, and molecular dynamics simulation.
2.1. Data Retrieval
The FASTA sequence of the human ARSB gene was obtained from UniProt using accession P15848. Missense mutation information was gathered through PubMed review and from dbSNP, HGMD, ClinVar, and Ensembl, with redundant nsSNPs removed.
The crystal structure of human ARSB was taken from the Protein Data Bank using PDB ID 1FSU. The text also states that remaining mutations were taken from Ensembl and that multiple mutation types were included during data collection.
- Sequence source: UniProt
- UniProt ID: P15848
- Structure source: Protein Data Bank
- PDB ID: 1FSU
- Variant sources listed: PubMed, dbSNP, HGMD, ClinVar, Ensembl
2.2. Sequence-Based Prediction of Deleterious Mutations
SIFT was used to predict functional impact based on sequence homology and conservation, with scores less than or equal to 0.05 treated as damaging. PolyPhen-2 evaluated substitutions using sequence and structure-based features and classified them as benign, possibly damaging, or probably damaging.
Mutation Assessor measured functional impact using evolutionary conservation patterns within protein families and subfamilies, and FATHMM applied hidden Markov model-based prediction with lower scores indicating higher pathogenic likelihood.
- Sequence-based tools listed: SIFT, PolyPhen-2, Mutation Assessor, FATHMM
- SIFT damaging threshold: ≤ 0.05
- Mutation Assessor deleterious FI threshold noted: > 2.0
2.3. Structure-Based Stability Prediction
MAESTROweb, mCSM, DynaMut2, and PremPS were used to predict mutation-induced stability changes. These tools estimate changes in Gibbs free energy (ΔΔG) and, in the case of DynaMut2, changes in vibrational entropy and conformational flexibility.
The paper explains that these methods help identify destabilizing substitutions and mutation hotspots by combining structural and evolutionary features with machine-learning or graph-based signatures.
- Structure-based tools listed: MAESTROweb, mCSM, DynaMut2, PremPS
- Negative ΔΔG is described in the methods as destabilizing for MAESTROweb and mCSM
- DynaMut2 also evaluates ΔΔS / flexibility changes
2.4. Pathogenicity Prediction Tools
MutPred2 was used as a machine-learning-based pathogenicity predictor that also suggests possible molecular consequences such as altered secondary structure or interaction changes. SNPs&GO and PhD-SNP were used to classify disease-associated versus neutral variants using sequence, Gene Ontology, and support-vector-machine approaches.
- Pathogenicity tools listed: MutPred2, SNPs&GO, PhD-SNP
- MutPred2 high-confidence pathogenicity threshold mentioned: > 0.589
- PhD-SNP disease threshold mentioned: > 0.5
2.5. Evolutionary Conservation Analysis
ConSurf was used to evaluate evolutionary conservation of amino acid residues using multiple sequence alignment and phylogenetic analysis. The methods describe scores from 1 for variable residues to 9 for highly conserved residues and note that disease-associated substitutions often occur at highly conserved positions.
- Conservation tool: ConSurf
- ConSurf score range described: 1 to 9
2.6. Aggregation Propensity Analysis
SODA was used to predict effects of mutations on solubility, disorder, secondary structure, and aggregation propensity. Arpeggio was used to analyze interatomic interactions such as hydrogen bonds, van der Waals contacts, hydrophobic interactions, and ionic interactions in protein structures.
- Aggregation tool: SODA
- Interaction-analysis tool: Arpeggio
4. Results and Discussion
The article combines results and discussion into a single major section. It evaluates ARSB variants stepwise using sequence, structure, disease-prediction, conservation, aggregation, interaction, and molecular-dynamics analyses to explain how selected mutations destabilize the protein and may contribute to MPS VI pathophysiology.
3.1. Deleterious Mutation Identification of nsSNPs
The paper states that sequence-based evaluation was carried out on ARSB nsSNPs using online tools including SIFT, PolyPhen2, PROVEAN, Mutation Assessor, and FATHMM, while structure-based analysis used PremPS, MAESTROweb, DynaMut2, and mCSM on variants located within the structurally resolved region.
The narrative reports that sequence-based analysis highlighted dangerous substitutions in counts of 302, 264, 238, and 167 depending on the tool, as shown graphically in Figure 2 on page 4. Structure-based predictions identified destabilizing substitutions with counts 132, 130, 64, and 130 across mCSM, DynaMut2, MAESTROweb, and PremPS, as summarized in Figure 3 on page 4.
After intersecting sequence-based and structure-based outputs, the article says that 44 amino acid changes were identified as harmful and destabilizing candidates for further investigation.
- Sequence-based tools named in results: SIFT, PolyPhen2, PROVEAN, Mutation Assessor, FATHMM
- Figure 2 on page 4 visualizes sequence-based deleterious mutation counts
- Figure 3 on page 4 visualizes structure-based destabilizing versus stabilizing counts
- Final harmful/destabilizing set highlighted in this subsection: 44 mutations
3.2. Identification of Pathogenic Mutations Using a Computational Approach
The disease-phenotype step used PhD-SNP, SNPs&GO, and MutPred2. The text says that among 57 high-confidence variants identified through the earlier sequence and structure workflow, 44 were classified as pathogenic after disease-phenotype assessment.
The article explicitly lists the 44 pathogenic mutations as A237D, C91R, E323K, E483D, G149R, G302R, G324V, G56D, G64R, H393P, I296N, I67N, K145E, L129P, L236P, L360P, L498P, L51P, L72P, L72R, L82P, L82R, L90P, L98P, L98R, P93R, R315P, R315Q, R327G, R327Q, R388T, T92K, V277G, V80G, W146R, W146S, W353R, W438G, Y138C, Y175D, Y210C, Y266S, Y86C, and Y86N.
Table 3 on page 8 provides per-variant disease predictions from PhD-SNP, SNPs&GO, and MutPred2 and also includes a final remark column.
- High-confidence variants before phenotype classification: 57
- Pathogenic variants after phenotype classification: 44
- Table 3 location: page 8
3.3. Analysis of Conserved Residue
ConSurf analysis was used to assess evolutionary conservation across the ARSB protein. According to the text, 12 of the final mutations were considered especially disease-causing because they fell within highly conserved regions.
The 12 highlighted conserved-region variants are A237D, G56D, G64R, L51P, I67N, L236P, W353R, V277G, T92K, R315Q, R315P, and H393P. Figure 5 on page 5 presents the conserved-residue mapping across the ARSB region.
- Highly conserved disease-linked set highlighted: 12 mutations
- Figure 5 location: page 5
3.4. Analysis of Aggregation Propensity
SODA was used to estimate effects of selected mutations on solubility and aggregation. Table 1 on page 5 reports values for 12 variants and marks A237D and W353R as less soluble, while the remaining listed variants are marked as more soluble or unchanged.
The text surrounding Table 1 contains multiple count statements, including that 12 vnsSNPs decrease protein solubility, that all three solubility-reducing alterations are concerning, and that 29 nsSNPs enhanced amino-acid dissolution. These statements do not fully align with the table itself, so they are preserved as extraction notes rather than normalized.
Figure 6 on page 5 shows mutant structural snapshots for A237D and W353R and is used in the discussion to explain how altered noncovalent interactions may contribute to misfolding, loss of function, or aggregation.
- Table 1 less-soluble variants explicitly listed: A237D and W353R
- Table 1 more-soluble / non-negative entries listed: G56D, G64R, H393P, I67N, L236P, L51P, R315P, R315Q, T92K, V277G
- Figure 6 location: page 5
3.4–3.5. Structural Interaction Interpretation
Table 2 on page 6 compares noncovalent interactions in wild-type ARSB versus A237D and W353R mutants. For wild type, the table lists 71 proximal contacts plus 3 van der Waals clashes for a total of 74. A237D is reported with 1 van der Waals contact, 11 van der Waals clashes, and 119 proximal contacts for a total of 131, while W353R is reported with 4 van der Waals contacts, 7 van der Waals clashes, and 119 proximal contacts for a total of 130.
The text argues that A237D increases local contacts and structural strain but also gains polar, hydrogen-bond, and ionic interactions, potentially preserving some integrity. By contrast, W353R is described as locally destabilizing through altered aromatic-region interactions and reduced hydrophobic organization.
Figure 7 on page 6 presents a conceptual disease model in which ARSB mutation leads to accumulation of sulfated glycosaminoglycans in lysosomes and contributes to lysosomal disorder pathogenesis.
- Table 2 location: page 6
- Wild-type total contact count in Table 2: 74
- A237D total contact count in Table 2: 131
- W353R total contact count in Table 2: 130
- Figure 7 location: page 6
3.6. Molecular Dynamics Simulation Analysis
Selected mutant variants and wild-type ARSB were subjected to molecular dynamics simulation to validate structural consequences of high-confidence deleterious substitutions. The analysis included RMSD, RMSF, radius of gyration (Rg), SASA, and hydrogen-bond evaluations.
Compared with wild type, mutant proteins are described as showing delayed equilibration, higher RMSD, greater flexibility in conserved and functional regions, increased Rg and SASA, reduced compactness, and greater solvent exposure. The authors interpret these patterns as consistent with unfolding tendency and increased aggregation propensity.
Figure 8 on page 7 presents radius-of-gyration analysis, while Figure 9 on page 7 presents RMSD behavior and conformational deviation during the simulation.
- MD metrics named: RMSD, RMSF, Rg, SASA, hydrogen bonds
- Figure 8 location: page 7
- Figure 9 location: page 7
5. Conclusion
The conclusion states that SNPs are among the most common genetic variants linked to human disease and argues that computational mutational analysis can reveal mechanisms underlying MPS VI pathogenesis.
According to the conclusion text, 139 of 429 mutations were harmful and destabilizing by sequence- and structure-based study, 44 variants were harmful after pathogenicity assessment, and two final mutations remained as likely disease-driving factors after ConSurf and aggregation analysis.
The article presents the work as a foundation for future targeted therapy development and as evidence for the value of advanced computational analysis in understanding mutation-driven molecular pathology in ARSB.
- Conclusion-level harmful/destabilizing count reported: 139 of 429
- Conclusion-level final disease-driving mutations after conservation/aggregation narrowing: 2
- Therapeutic implication: identified mutations may guide targeted treatment strategies
6. Statements and Declarations
The article includes end-matter sections for conflicts of interest, funding, data availability, and declaration of AI-tool use.
Conflicts of Interest
The authors declare no conflict of interest.
Funding
This work received no funding.
Data availability statement
All data generated or analyzed during this study are included in this manuscript.
Declaration on the Use of AI Tools
The authors declare that ChatGPT (OpenAI) was used solely to refine the language, improve grammar, and enhance the clarity of the manuscript.
Figures
The PDF includes three numbered figures covering the annotation workflow and two sets of structural superimpositions for selected modeled proteins.
Figure 1

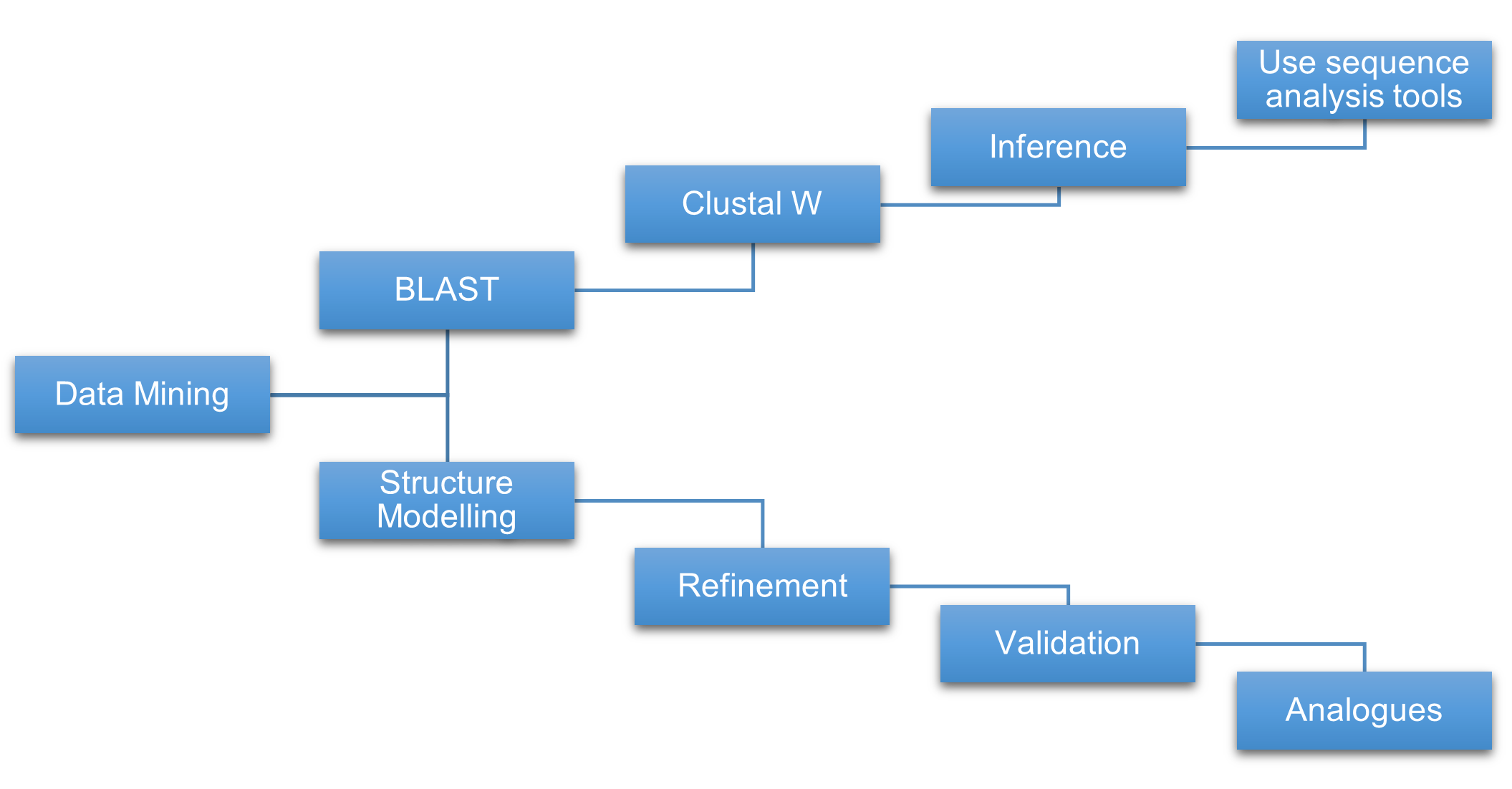{kind=link}
Figure 1. Diagram of the computational methods used to forecast pathogenicity of ARSB mutations across sequence-based, structure-based, disease-prediction, and sequence-aggregation analyses.
Download figure
Figure 2. Sequence-based deleterious mutation counts for ARSB shown as a computational summary graph.
Download figure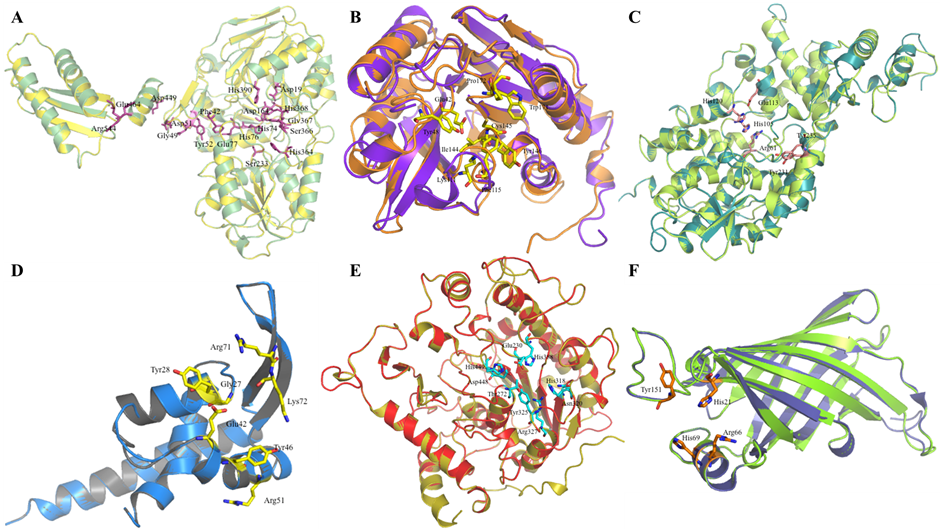{kind=link}

Figure 3. Structure-based deleterious mutation counts for ARSB, comparing destabilizing and stabilizing predictions across tools.
Download figure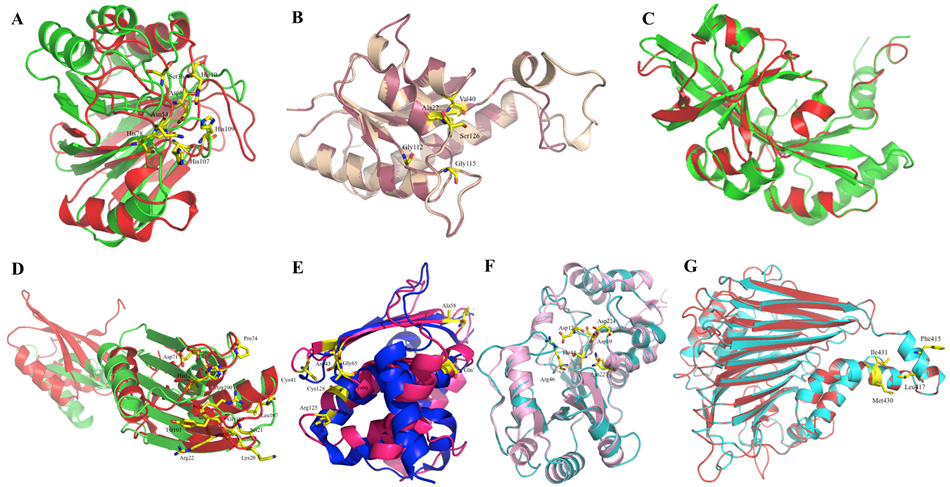{kind=link}

Figure 4. Pathogenic mutations predicted in the ARSB gene using structure-based tools and disease-prediction outputs.
Download figure{kind=link}
Figure 5. Conserved residue map of the ARSB region used for ConSurf-based conservation interpretation.
Figure 6. Mutant structural views for A237D and W353R illustrating local structural defects and altered interaction environments.
Figure 7. Conceptual model showing ARSB mutation-driven accumulation of sulfated glycosaminoglycans in lysosomes and resulting lysosomal disorder.
Figure 8. Radius of gyration analysis showing compactness changes over time in wild-type and mutant ARSB proteins.
Figure 9. RMSD plot showing structural stability and conformational deviation of wild-type and mutant ARSB proteins during simulation.
References
- 1
De Ponti, G., Donsante, S., Frigeni, M., et al. 2022, International Journal of Molecular Sciences, 23, 11168
- 2
Enni, M. A. 2025, International Journal of Scientific Interdisciplinary Research, 6, 88
- 3
Gomez-Ospina, N. 2024, Arylsulfatase A deficiency
- 4
Gros, F., & Muller, S. 2023, Nature Reviews Nephrology, 19, 366
- 5
Leal, A. F., Prieto, L. E., Pachajoa, H., & Tomatsu, S. 2025, Molecular Genetics and Metabolism, 109255
- 6
Lipiński, P., Różdźyńska-Świątkowska, A., Wiśniewska, K., et al. 2025, Biomolecules, 15, 1448
- 7
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F., & Tifft, C. J. 2018, Nature Reviews Disease Primers, 4, 27
- 8
Rossi, A., Romano, R., Fecarotta, S., et al. 2025, Med, 6
- 9
Sarachakov, A., Yudina, A., Svekolkin, V., et al. 2025, Human Genetics, 144, 1245
- 10
Sauna, Z. E., & Kimchi-Sarfaty, C. 2022, Single nucleotide polymorphisms: human variation and a coming revolution in biology and medicine (Springer)
- 11
Scerra, G., De Pasquale, V., Scarcella, M., et al. 2022, Open Biology, 12
- 12
Sinha, A., Dinakarkumar, Y., Al-Qahtani, W. H., et al. 2022, Human Gene, 34, 201079
- 13
Tobacman, J. K., & Bhattacharyya, S. 2022, International Journal of Molecular Sciences, 23, 13146
Tables
Table 1. List of hypothetical proteins in Listeria monocytogenes (Part 1 of 3).
| S. No. | Name of Hypothetical Protein | pI | Length | Pfam | Proposed Function |
|---|---|---|---|---|---|
| 1 | C1L300/ C1L300_LISMC | 9.69 | 447 | MatE (PF01554) | antiporter activity, drug transmembrane transporter activity |
| 2 | C1L2X5/ C1L2X5_LISMC | 7.85 | 345 | UPF0118 (PF01594) | NA |
| 3 | C1L2V8/ C1L2V8_LISMC | 5.02 | 120 | DUF964 (PF06133) | NA |
| 4 | C1L2V7/ C1L2V7_LISMC | 5.45 | 267 | YmdB (PF13277) | putative phosphoesterases |
| 5 | C1L2S2/ C1L2S2_LISMC | 6.41 | 274 | FtsJ (PF01728), S4 (PF01479) | methyltransferase involved in viral RNA capping, RNA binding |
| 6 | C1L2Q9/ C1L2Q9_LISMC | 6.47 | 321 | DUF1385 (PF07136) | NA |
| 7 | C1L2P0/ C1L2P0_LISMC | 4.78 | 118 | No domains | NA |
| 8 | C1L2N2/ C1L2N2_LISMC | 5.44 | 92 | DUF503 (PF04456) | NA |
| 9 | C1L2K8/ C1L2K8_LISMC | 5.38 | 174 | Acetyltransf_3 (PF13302) | transfers acetyl group |
| 10 | C1L2J0/ C1L2J0_LISMC | 7.84 | 103 | NA | NA |
| 11 | C1L2E5/ C1L2E5_LISMC | 5.13 | 174 | Metallophos_2 (PF12850) | Phosphoesterase (hydrolase activity, acting on ester bonds) |
| 12 | C1L1Z7/ C1L1Z7_LISMC | 6.42 | 774 | Glycos_transf_2 (PF00535), Glyphos_transf (PF04464) | glycerophosphotransferase (sequential transfer of glycerol-phosphate units) (CDP-glycerol glycerophosphotransferase activity) |
| 13 | C1L1V8/ C1L1V8_LISMC | 9.43 | 267 | DUF817 (PF05675) | NA |
| 14 | C1L1U8/ C1L1U8_LISMC | 6.18 | 555 | Lactamase_B (PF00753), RMMBL_DRMBL (CL0398) | RNA binding, hydrolase activity, acting on ester bonds, metal ion binding |
| 15 | C1L1S5/ C1L1S5_LISMC | 10.1 | 344 | DUF218 (PF02698) | NA |
| 16 | C1L1R3/ C1L1R3_LISMC | 9.3 | 255 | TerC (PF03741) | integral to membrane |
| 17 | C1L1R2/ C1L1R2_LISMC | 9.36 | 255 | TerC (PF03741) | integral to membrane |
| 18 | C1L1R1/ C1L1R1_LISMC | 9.09 | 453 | MatE (PF01554) | antiporter activity, drug transmembrane transporter activity |
| 19 | C1L1K4/ C1L1K4_LISMC | 9.81 | 201 | SNARE_assoc (PF09335) | NA |
| 20 | C1L1G8/ C1L1G8_LISMC | 5.59 | 725 | Tex_N (PF09371), HHH_3 (PF12836), S1 (PF00575) | RNA binding, hydrolase activity- acting on ester bonds |
| 21 | C1L1B3/ C1L1B3_LISMC | 4.78 | 125 | Ribonuc_L-PSP (PF01042) | Inhibits protein synthesis by cleaving the mRNA |
| 22 | C1L190/ C1L190_LISMC | 5.09 | 282 | NA | NA |
| 23 | C1L165/ C1L165_LISMC | 4.69 | 176 | YceI (PF04264) | binds to polyisoprenoid |
| 24 | C1L164/ C1L164_LISMC | 9.54 | 306 | EamA (PF00892) | transporter activity |
| 25 | C1L162/ C1L162_LISMC | 9.27 | 233 | DUF554 (PF04474) | NA |
| 26 | C1L161/ C1L161_LISMC | 4.79 | 296 | CN_hydrolase (PF00795) | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds |
| 27 | C1L158/ C1L158_LISMC | 5.68 | 282 | PhzC-PhzF (PF02567) | Phenazine biosynthesis |
| 28 | C1L143/ C1L143_LISMC | 5.33 | 309 | DAGK_cat (PF00781) | NAD+ kinase activity, diacylglycerol kinase activity |
| 29 | C1L0U3/ C1L0U3_LISMC | 4.83 | 288 | Hydrolase_3 (PF08282) | hydrolase activity |
| 30 | C1L0T5/ C1L0T5_LISMC | 5.42 | 209 | HhH-GPD (PF00730) | DNA binding, endonuclease activity |
| 31 | C1L0T0/ C1L0T0_LISMC | 9.82 | 187 | DUF420 (PF04238) | NA |
Table 2. List of hypothetical proteins in Listeria monocytogenes (Part 2 of 3).
| S. No. | Name of Hypothetical Protein | pI | Length | Pfam | Proposed Function |
|---|---|---|---|---|---|
| 32 | C1L0R8/ C1L0R8_LISMC | 5.34 | 606 | Sulfatase (PF00884) | sulfuric ester hydrolase activity |
| 33 | C1L0Q0/ C1L0Q0_LISMC | 9.71 | 246 | TauE (PF01925) | NA |
| 34 | C1L0P3/ C1L0P3_LISMC | 4.87 | 172 | Acetyltransf_1 (PF00583) | acetyltransferase activity |
| 35 | C1L0M8/ C1L0M8_LISMC | 4.78 | 110 | PadR (PF03551) | involved in negative regulation of phenolic acid metabolism |
| 36 | C1L0L0/ C1L0L0_LISMC | 6.08 | 394 | Methyltrans_SAM (PF10672) | methyltransferase activity |
| 37 | C1L0K2/ C1L0K2_LISMC | 7.7 | 431 | Xan_ur_permease (PF00860) | transporter activity |
| 38 | C1L0G9/ C1L0G9_LISMC | 4.82 | 264 | AP_endonuc_2 (PF01261) | involved in the myo-inositol catabolism (isomerase activity) |
| 39 | C1L075/ C1L075_LISMC | 5.18 | 346 | Lactonase (PF10282) | 6-phosphogluconolactonase activity |
| 40 | C1L028/ C1L028_LISMC | 5.72 | 143 | Usp (PF00582) | response to stress |
| 41 | C1L024/ C1L024_LISMC | 5.26 | 226 | GATase (PF00117) | transferase activity |
| 42 | C1KZV6/ C1KZV6_LISMC | 4.89 | 281 | NmrA (PF05368) | nucleotide binding (negative transcriptional regulator involved in the post-translational modification of the transcription factor AreA.) |
| 43 | C1KZU1/ C1KZU1_LISMC | 5.41 | 377 | Gly_kinase (PF02595) | glycerate kinase activity |
| 44 | C1KZQ3/ C1KZQ3_LISMC | 4.64 | 279 | Hydrolase_3 (PF08282) | hydrolase activity |
| 45 | C1KZM7/ C1KZM7_LISMC | 8.93 | 156 | Usp (PF00582) | response to stress |
| 46 | C1KZM4/ C1KZM4_LISMC | 6.15 | 120 | YjbR (PF04237) | NA |
| 47 | C1KZE1/ C1KZE1_LISMC | 4.83 | 270 | Hydrolase_3 (PF08282) | hydrolase activity |
| 48 | C1KZ86/ C1KZ86_LISMC | 5.18 | 111 | PhnA_Zn_Ribbon (PF08274), PhnA (PF03831) | NA |
| 49 | C1KZ81/ C1KZ81_LISMC | 5.97 | 494 | FTR1 (PF03239) | transmembrane transport |
| 50 | C1KZ17/ C1KZ17_LISMC | 8.78 | 220 | MgtC (PF02308) | transport of Mg2+ |
| 51 | C1KZ02/ C1KZ02_LISMC | 5.31 | 280 | SAM_adeno_trans (PF01887) | NA |
| 52 | C1KYZ6/ C1KYZ6_LISMC | 8.7 | 362 | MacB_PCD (PF12704), FtsX (PF02687) | transport lipids targeted to the outer membrane across the inner membrane |
| 53 | C1KYZ4/ C1KYZ4_LISMC | 5.44 | 97 | ABM (PF03992) | monooxygenase activity |
| 54 | C1KYZ2/ C1KYZ2_LISMC | 4.62 | 275 | Hydrolase_3 (PF08282) | hydrolase activity |
| 55 | C1KYY1/ C1KYY1_LISMC | 5.4 | 440 | HD (PF01966) | metal ion binding, phosphoric diester hydrolase activity |
| 56 | C1KYX9/ C1KYX9_LISMC | 6.83 | 217 | Peptidase_M50 (PF02163) | proteolysis, metalloendopeptidase activity |
| 57 | C1KYX3/ C1KYX3_LISMC | 9.03 | 306 | DAGK_cat (PF00781) | kinase, transferase |
| 58 | C1KYW9/ C1KYW9_LISMC | 10.1 | 357 | UPF0104 (PF03706) | NA |
| 59 | C1KYT3/ C1KYT3_LISMC | 5.78 | 211 | UPF0029 (PF01205), DUF1949 (PF09186) | GTP binding |
| 60 | C1KYS5/ C1KYS5_LISMC | 6.43 | 287 | DUF161 (PF02588), DUF2179 (PF10035) | NA |
| 61 | C1KYP0/ C1KYP0_LISMC | 4.97 | 322 | UPF0052 (PF01933) | NA |
| 62 | C1KYM2/ C1KYM2_LISMC | 5.12 | 259 | CN_hydrolase (PF00795) | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds |
Table 3. List of hypothetical proteins in Listeria monocytogenes (Part 3 of 3).
| S. No. | Name of Hypothetical Protein | pI | Length | Pfam | Proposed Function |
|---|---|---|---|---|---|
| 63 | C1KYL6/ C1KYL6_LISMC | 5.39 | 273 | Hydrolase_3 (PF08282) | ATP binding, ATPase activity, coupled to transmembrane movement of ions, phosphorylative mechanism |
| 64 | C1KYI4/ C1KYI4_LISMC | 5.98 | 201 | MTS (PF05175) | 16S rRNA (guanine(966)-N(2))-methyltransferase activity |
| 65 | C1KY64/ C1KY64_LISMC | 5.76 | 117 | ArsC (PF03960) | regulate the transcription of multiple genes in response to disulfide stress |
| 66 | C1KY61/ C1KY61_LISMC | 5.27 | 291 | Cation_efflux (PF01545) | cation transmembrane transporter activity |
| 67 | C1KY51/ C1KY51_LISMC | 5.94 | 147 | NifU_N (PF01592) | iron ion binding, iron-sulfur cluster binding |
| 68 | C1KY50/ C1KY50_LISMC | 4.87 | 464 | UPF0051 (PF01458) | iron-sulfur cluster assembly |
| 69 | C1KY46/ C1KY46_LISMC | 9.02 | 279 | TauE (PF01925) | involved in the transport of anions across the cytoplasmic membrane during taurine metabolism as an exporter of sulfoacetate |
| 70 | C1KY45/ C1KY45_LISMC | 5.08 | 462 | Metallophos (PF00149), 5_nucleotid_C (PF02872) | hydrolase activity (5_nucleotid_C) |
| 71 | C1KY43/ C1KY43_LISMC | 4.94 | 255 | Hydrolase_like (PF13242), Hydrolase_6 (PF13344) | hydrolase activity |
| 72 | C1KY41/ C1KY41_LISMC | 4.82 | 436 | DUF21 (PF01595), CBS (PF00571), CorC_HlyC (PF03471) | flavin adenine dinucleotide binding (CBS) |
| 73 | C1KY30/ C1KY30_LISMC | 9.43 | 406 | Voltage_CLC (PF00654) | voltage-gated chloride channel activity |
| 74 | C1KY03/ C1KY03_LISMC | 5.29 | 156 | Rrf2 (PF02082) | NA |
| 75 | C1KXZ5/ C1KXZ5_LISMC | 4.65 | 281 | Hydrolase_3 (PF08282) | hydrolase activity |
| 76 | C1KXZ0/ C1KXZ0_LISMC | 4.76 | 532 | Amidohydro_3 (PF07969) | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in cyclic amides |
| 77 | C1KXY4/ C1KXY4_LISMC | 6.26 | 257 | Methyltransf_26 (PF13659) | methyltransferase activity |
| 78 | C1KXX7/ C1KXX7_LISMC | 4.83 | 270 | Hydrolase_3 (PF08282) | hydrolase activity |
| 79 | C1KXV5/ C1KXV5_LISMC | 5.52 | 249 | EAL (PF00563) | NA |
| 80 | C1KXN7/ C1KXN7_LISMC | 6.53 | 331 | Bac_luciferase (PF00296) | oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen |
| 81 | C1KXN1/ C1KXN1_LISMC | 6.58 | 147 | DUF523 (PF04463) | NA |
| 82 | C1KWJ0/ C1KWJ0_LISMC | 6.74 | 121 | DUF1798 (PF08807) | NA |
| 83 | C1KWG8/ C1KWG8_LISMC | 6.52 | 327 | DUF939 (PF06081), DUF939_C (PF11728) | NA |
| 84 | C1KWG7/ C1KWG7_LISMC | 4.39 | 125 | Glyoxalase_2 (PF12681) | NA |
| 85 | C1KWG4/ C1KWG4_LISMC | 6.52 | 205 | HTH_11 (PF08279), CBS (PF00571) | sequence-specific DNA binding transcription factor activity (CBS) |
| 86 | C1KWC7/ C1KWC7_LISMC | 5.29 | 291 | YicC_N (PF03755), DUF1732 (PF08340), DUF1732 (PF08340) | play a role in the stationary phase survival (YicC) |
| 87 | C1KW03/ C1KW03_LISMC | 6.11 | 126 | OsmC (PF02566) | has a novel pattern of oxidative stress regulation |
| 88 | C1KVW1/ C1KVW1_LISMC | 6.22 | 192 | rRNA_methylase (PF06962) | methyltransferase activity |
| 89 | C1KVW0/ C1KVW0_LISMC | 5.48 | 321 | Radical_SAM (PF04055) | catalytic activity, iron-sulfur cluster binding |
| 90 | C1KVA0/ C1KVA0_LISMC | 6.02 | 234 | DUF633 (PF04816) | tRNA (adenine-N1-)-methyltransferase activity |
| 91 | C1KV99/ C1KV99_LISMC | 5.5 | 373 | NIF3 (PF01784) | NIF3 interacts with the yeast transcriptional coactivator NGG1p, which is part of the ADA complex |
| 92 | C1KV71/ C1KV71_LISMC | 4.92 | 269 | Hydrolase_3 (PF08282) | cation transport, ATP binding, ATPase activity, coupled to transmembrane movement of ions, phosphorylative mechanism |
Table 4. Major Classes of Hypothetical Proteins in Listeria monocytogenes
| S. No. | Proposed function of Hypothetical protein | Primary Accession number |
|---|---|---|
| 1 | Hydrolase | C1L0U3, C1L0R8, C1KZQ3, C1KZE1, C1KYZ2, C1KYY1, C1KY45, C1KY43, C1KXZ5 |
| 2 | Transferase | C1L2K8, C1L1Z7, C1L0P3, C1L0L0, C1L024, C1KYI4, C1KXY4, C1KVW1, C1KVA0 |
| 3 | Transporter | C1L300, C1L1R1, C1L164, C1L0K2, C1KZ81, C1KZ17, C1KYZ6, C1KY61, C1KY46 |
| 4 | RNA binding | C1L2S2, C1L1U8, C1L1G8 |
| 5 | Hydrolase acting on carbon-nitrogen but not on peptide | C1L161, C1KYM2, C1KXZ0 |
| 6 | Kinase | C1L143, C1KZU1, C1KYX3 |
| 7 | Response to stress | C1LO28, C1KZM7, C1KW03 |
| 8 | Integral membrane | C1L1R3, C1L1R2, C1KY64 |
| 9 | DNA binding | C1L0T5, C1KWG4 |
| 10 | Phosphoesterase | C1L2V7, C1L2E5 |
| 11 | ATP Binding | C1KYL6, C1KV71 |
| 12 | Others | C1L1B3, C1L165, C1L158, C1L0M8, C1L0G9, C1L075, C1KZV6, C1KYZ4, C1KYX9, C1KYT3, C1KY51, C1KY50, C1KY41, C1KY30, C1KXN7, C1KWC7, C1KVW8, C1KV99 |
| 13 | Unknown | C1L2X5, C1L2V8, C1L2Q9, C1L2P0, C1L2J0, C1L1S5, C1L1K4, C1L198, C1L162, C1L0T0, C1L0Q0, C1KZM4, C1KZ86, C1KZ02, C1KYW9, C1KYS5, C1KYP0, C1KY03, C1KYN7, C1KXN1, C1KWJ0, C1KWG8, C1KWG7 |
Table 5. Superimposed residues between template and modeled proteins C1L1U8–C1L165.
| Protein ID | Superimposed Residues (Template → Model) |
|---|---|
| C1L1U8 | Asp78→Asp78, His79→His79, Asp164→Asp164, His390→His390, His74→His74, His76→His76, His142→His142, Asp195→Asp195, His368→His368, Gly49→Gly49, Asp51→Asp51, Asp443→Asp443, Glu464→Glu464, Asp449→Asp449, Arg544→Arg544, Ser366→Ser366, His364→His364, Gly367→Gly367, Ser233→Ser233, Glu77→Glu77, Phe42→Phe42, Tyr52→Tyr52 |
| C1KYM2 | Glu43→Glu42, Lys109→Lys111, Cys143→Cys145, Tyr144→Tyr146, Phe49→Tyr48, Phe113→Phe115, Trp175→Trp171, Val142→Ile144, Pro176→Pro172 |
| C1KYY1 | His129→His120, Glu122→Glu113, His66→His64, His110→His110, Asp111→Asp102, Asp183→Asp173, Lys14→Lys12, Asn36→Ala34, Gln41→Gln39, Arg326→Arg317, Lys330→Arg321, His114→His105, Tyr239→Tyr231, Arg63→Arg61, Leu49→Leu47, His119→His110, Tyr187→Tyr177, Tyr243→Tyr235, Tyr368→Tyr358 |
| C1L0M8 | Gly25→Gly27, Tyr26→Tyr28, Glu42→Glu42, Arg71→Arg71, Lys72→Lys72, Tyr46→Tyr46, Arg51→Arg51 |
| C1L0R8 | Thr297→Thr272, His412→His388, Glu253→Glu230, Trp350→Tyr325, His472→His449, Asp471→Asp448, His343→His318, Asn345→Asn320, Arg352→Arg327, Thr408→Ser384 |
| C1L165 | His18→His21, Arg62→Arg66, His65→His69, Trp146→Tyr151 |
Table 6. Superimposed residues between template and modeled proteins C1L2E5–C1KY50.
| Protein ID | Superimposed Residues (Template → Model) |
|---|---|
| C1L2E5 | Asp8→Asp8, His10→His10, Asp36→Ser36, Asn59→Asn54, Asn60→Cys55, His97→His78, His120→His107, Thr121→Ser108, His122→His109 |
| C1KZM7 | Arg135→Tyr130, Ser131→Ser126, Val38→Val40, Pro8→Gly10, Gly117→Gly112, Gln119→Thr114, Ala133→Ser128, Val132→Val127, Gly120→Gly115, Gly123→Ala118, Asn122→Ser117 |
| C1KYZ6 | Leu469→Lys186, Tyr465→Trp182, Asn346→Ala79, Ile342→Thr75 |
| C1KYT3 | Ser23→Ser21, His54→His52, Glu77→Glu74, Arg104→Arg100, Lys22→Lys20, Arg24→Arg22, Phe25→Phe23, Asp75→Asp71, Gly76→Gly72, Pro78→Pro74, Ala82→Ala78, Tyr105→Tyr101, Tyr106→Phe102, Gly107→Gly103, Leu111→Leu107, Leu116→Leu112, Tyr120→Tyr116, Asp74→Asp70, Thr81→Thr77 |
| C1KY51 | Cys40→Cys41, Asp42→Asp43, Cys65→Cys66, Arg124→Arg125, Cys127→Cys128, Gly64→Gly65, Asp57→Ala58, Asp77→Gln78, Glu56→Val57 |
| C1KYL6 | Trp171→Ser187, Phe175→Asn191, Arg45→Arg46, Asp1→Asp12, Thr43→Thr44, Asp8→Asp10, Lys185→Lys201, Asn211→Asn227, Asp208→Asp22 |
| C1KY50 | Phe373→Phe415, Leu375→Leu417, Ile380→Leu422, Met338→Met430, Ile389→Ile431, Ala392→Gly434, Ala395→Glu437 |