


About Article
Impact of Single Nucleotide Polymorphisms in TERF1-Interacting Nuclear Factor 2 and Their Association with Dyskeratosis Congenita Autosomal Dominant-3
Abstract
Dyskeratosis congenita (DC) is a rare hereditary telomere biology disorder characterized by bone marrow failure, nail dystrophy, mucosal leukoplakia, and abnormal skin pigmentation. Telomere shortening resulting from defects in telomere maintenance genes is a central pathogenic mechanism of the disease. TERF1-interacting nuclear factor 2 (TIN2), encoded by the TINF2 gene, is a critical component of the shelterin complex that safeguards telomere integrity by coordinating interactions among TRF1, TRF2, and TPP1/POT1. Heterozygous missense mutations in TINF2 are known to cause autosomal dominant dyskeratosis congenita type 3. In this study, a comprehensive in silico analysis was performed to identify deleterious and pathogenic non-synonymous single-nucleotide polymorphisms (nsSNPs) in the TIN2 protein. A total of 271 nsSNPs were subjected to sequence-based prediction tools (SIFT, PROVEAN, PolyPhen-2, Mutation Assessor, and SAAFEC-SEQ). Structure-based stability analyses of 134 variants located within the resolved crystal structure (PDB ID: 5XYF) were conducted using SDM2, DUET, mCSM, and MUpro. High-confidence deleterious and destabilizing mutations were further evaluated for disease association using PMut, PhD-SNP, and Rhapsody. Twelve missense mutations (A16D, V22G, V34G, L38P, V49G, R52P, R56G, E109G, F114S, L121P, Y139C, and L150P) were consistently predicted as pathogenic. Detailed structural characterization revealed significant alterations in protein stability, packing density, accessible surface area, aggregation propensity, and intermolecular noncovalent interactions. Most of these mutations showed increased aggregation tendency or reduced solubility, suggesting disruption of shelterin complex stability. This study provides structural and functional insights into pathogenic TINF2 variants and highlights potential molecular mechanisms underlying autosomal dominant dyskeratosis congenita.
Keywords
1. Introduction
Telomeres are nucleoprotein complexes located at the linear ends of chromosomes that maintain genome stability and integrity. Human telomeres consist of approximately 5–15 kb of TTAGGG tandem repeats, forming double-stranded DNA with a 3' single-stranded overhang bound by the shelterin complex (Pike et al., 2019). The shelterin complex is composed of six subunits: TRF1, TRF2, POT1, TIN2, TPP1, and RAP1 (de Lange, 2005). This complex plays a critical role in protecting chromosome ends and regulating telomerase activity to maintain telomere length.
TIN2, encoded by the TINF2 gene, is a central component of the shelterin complex, facilitating its assembly by bridging the double-stranded telomeric DNA-binding proteins TRF1 and TRF2 with the single-stranded DNA-binding complex TPP1/POT1. Structurally, TIN2 contains a telomeric repeat factor homology (TRFH) domain that mediates essential protein–protein interactions within the complex. The N-terminal region of TIN2 exhibits structural similarity to the TRFH domains of TERF1 and TRF2, suggesting an evolutionary relationship, although these proteins have diverged functionally.
The TIN2 protein consists of 354 amino acids. Within the shelterin complex, TIN2 interacts with TPP1 through its N-terminal domain (approximately 200 amino acids) and contains a TRF1-binding motif spanning approximately 20 amino acids (Yang et al., 2011). Due to its central scaffolding role, TIN2 is essential for telomere length regulation and chromosome end protection. It contributes to TRF1-dependent telomere length control and stabilizes TRF1 through multiple mechanisms. Notably, TIN2 protects TRF1 from tankyrase 1-induced poly(ADP-ribosyl)ation, thereby preventing its dissociation from telomeres. Additionally, TIN2 competes with SCFFBX4 for TRF1 binding, protecting it from ubiquitin-dependent proteolysis (Frescas & de Lange, 2014).
Defects in telomere maintenance result in progressive telomere shortening and genomic instability, which are hallmarks of a group of inherited disorders known as telomeropathies. Dyskeratosis congenita (DC) is a rare hereditary bone marrow failure syndrome characterized by the classical triad of nail dystrophy, mucosal leukoplakia, and abnormal skin pigmentation (Savage et al., 2008). Patients with DC often develop additional complications, including aplastic anemia, pulmonary fibrosis, myelodysplastic syndrome, and leukemia. The disorder exhibits genetic heterogeneity and can be inherited in X-linked recessive, autosomal recessive, or autosomal dominant patterns. Mutations in several telomere-associated genes, including DKC1, TERC, TERT, NOP10, NHP2, TCAB1, RTEL1, and TINF2, have been implicated in DC. Notably, heterozygous missense mutations in TINF2 are responsible for autosomal dominant dyskeratosis congenita type 3 and are frequently associated with severe clinical manifestations and markedly shortened telomeres.
Several studies have identified mutation hotspots within residues 269–298 of TIN2, particularly near the TRF1-binding domain. These mutations are believed to disrupt shelterin complex assembly and compromise telomere stability. Given the critical scaffolding function of TIN2, even minor amino acid substitutions may significantly affect protein stability, intermolecular interactions, and overall telomere function. Non-synonymous single-nucleotide polymorphisms (nsSNPs) can alter protein structure and function, contributing to disease pathogenesis. Computational prediction tools provide a cost-effective and systematic approach to identify and prioritize potentially deleterious variants. Integrating sequence-based, structure-based, and pathogenicity prediction methods enables the identification of high-confidence disease-associated mutations and facilitates understanding of their molecular mechanisms.
In this study, we performed a comprehensive in silico analysis of nsSNPs in the TINF2 gene to identify deleterious, destabilizing, and pathogenic variants associated with dyskeratosis congenita. By integrating multiple predictive algorithms and structural analyses, we aimed to elucidate the molecular effects of selected mutations on TIN2 stability and function, thereby providing insights into the structural basis of autosomal dominant dyskeratosis congenita.
2. Materials and Methods
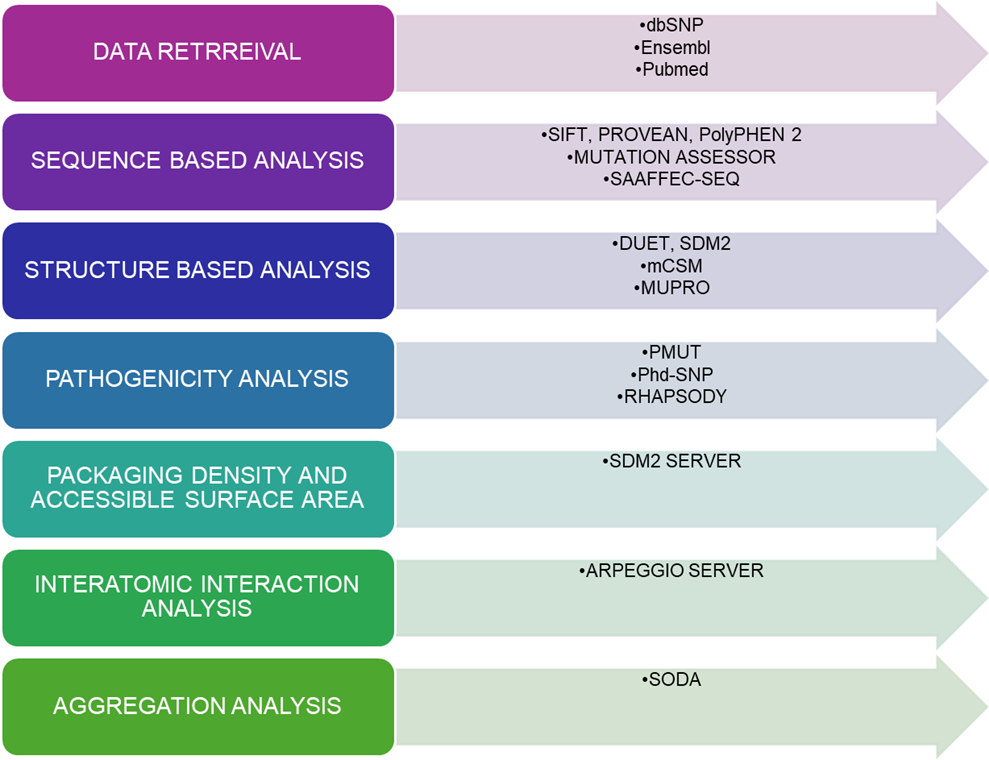
2.1. Data Retrieval
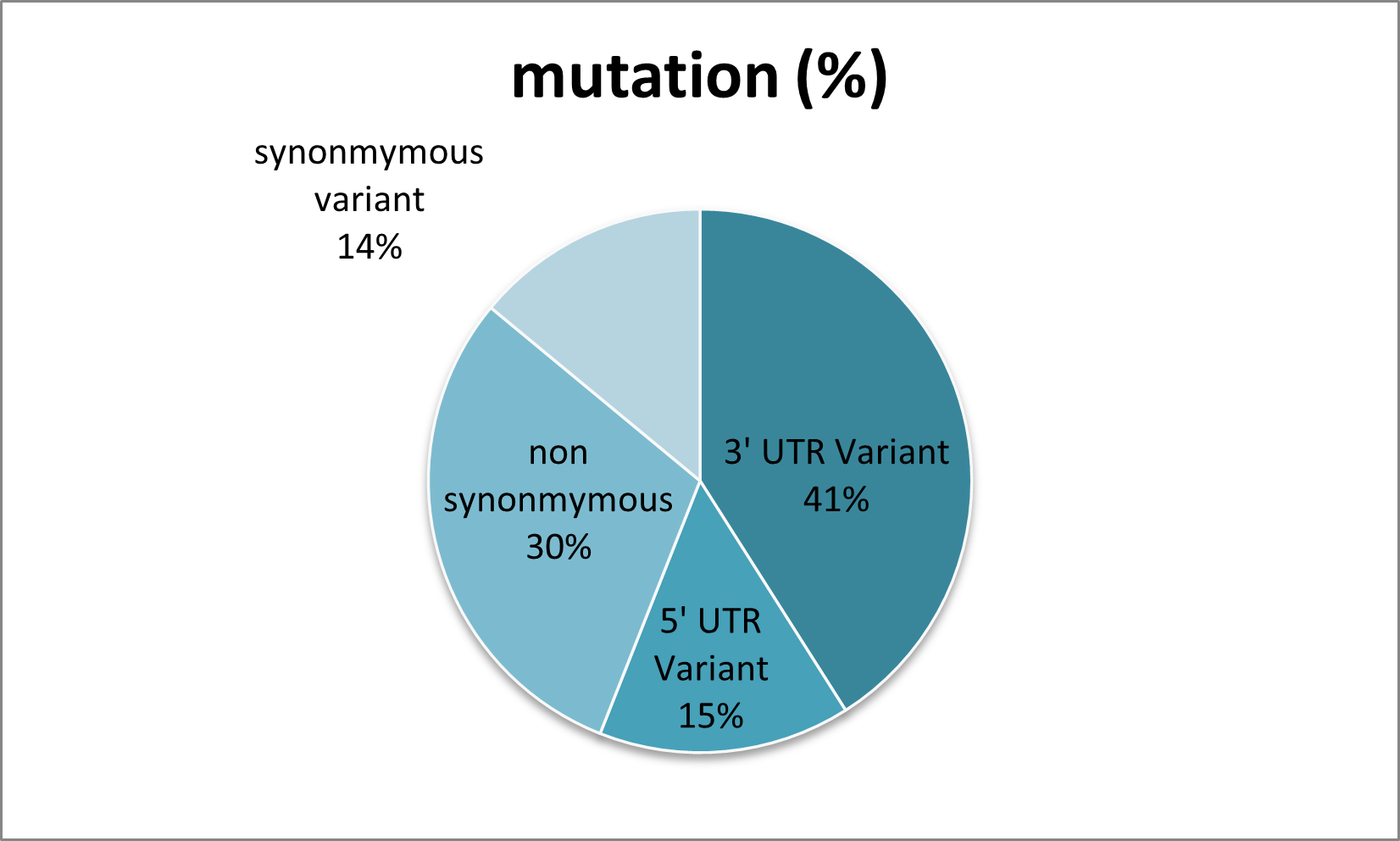
The FASTA format sequence of TINF2 protein was extracted from the UniProt database using UniProt ID (Q9BSI4). Using different databases such as dbSNP (Sherry et al., 2001), HGMD (Stenson et al., 2009), Clinvar (Landrum et al., 2014), and Ensembl (Hubbard et al., 2002) make a list of missense mutations and remove the duplicate nsSNPs. Approximately 900 mutations are obtained from the Ensembl database, of which 271 are non-synonymous mutations. The 3-D crystal structure obtained from the PDB (Berman et al., 2002) database of TINF2 protein using (PDB ID: 5XYF). This structure was used for structural-based analysis.
2.2. Prediction of deleterious mutations using sequence-based tools
SIFT The Sorting Intolerant from Tolerant (SIFT) program (http://sift.jcvi.org/) uses physical properties of amino acids to predict whether amino acid substitutions are deleterious or neutral. SIFT also estimates a protein’s sequence homology. If the SIFT score is more than 0.05, then the mutation is tolerable or neutral; if the score is equal to or less than 0.05, then it is not tolerable or deleterious (Kumar et al., 2009). All 271 nsSNPs mutations of the TINF2 protein are tested using the SIFT tool.
PolyPhen-2 Polymorphism phenotyping (PolyPhen-2) (http://genetics.bwh.harvard.edu/pph2/) is a sequence-based tool that finds the damaging probability of amino acid substitution in any protein on the basis of its physical and comparative properties (Ramensky et al., 2002). It takes a FASTA format sequence as input and estimates the PSIC score between 0 to 1. PolyPhen-2 scores range from 0.0 to 1.0. Variants closer to 1.0 are more likely to be damaging and are classified as benign, possibly damaging, or probably damaging based on predefined thresholds.
PROVEAN Protein variation effect analyzer (PROVEAN) (http://provean.jcvi.org/) is a sequence-based tool take FASTA format sequence as an input and predicts the damaging nsSNP of protein on the basis of the functionality of the protein (Choi & Chan, 2015). If the PROVEAN score is greater than -2.5, the mutation is predicted to be neutral; scores less than -2.5 are considered deleterious.
Mutation Assessor Mutation Assessor (http://mutationassessor.org/r3/) is a sequence-based tool that takes a UniProt ID or NCBI RefSeq ID as input for a protein. The mutation assessor classifies mutations as medium, low, or neutral in terms of deleteriousness based on multiple sequence alignment and evolutionary conservation (Reva et al., 2011). This tool predicts the functional impact of a missense mutation on a protein and computes the FI score. If the FI score is less than 2.00, the mutation is neutral; if the FI score is greater than 2.00, the mutation is considered deleterious.
SAAFEC-SEQ Single Amino Acid Folding free Energy Changes (SAAFEC-SEQ) (http://compbio.clemson.edu/SAAFEC-SEQ/index.php). SAAFSEC-SEQ is a sequence-based tool used to determine the change in folding free energy caused by amino acid substitution (Getov et al., 2016). It takes a protein FASTA sequence as input and predicts changes in free energy based on physicochemical properties and sequence features.
2.3. Prediction of the destabilizing mutation using structure-based tools
SDM2 Site-Directed Mutator (SDM2) (http://marid.bioc.cam.ac.uk/sdm2) is structure based tool that takes a PDB ID or a PDB file as input and uses environment-specific amino acid substitution tables to calculate the stability of an nsSNP in a protein (Pandurangan et al., 2017). It estimates the change in stability between the wild-type and mutant nsSNP of a protein using ΔΔ G. SDM2 estimates the change in stability (ΔΔ G) between the wild-type and mutant protein. Negative ΔΔ G values indicate destabilizing mutations, whereas positive values indicate stabilizing effects. All 134 nsSNPs located within the resolved crystal structure were analyzed using SDM2.
mCSM It is a structure-based tool to estimate nsSNP. mCSM take PDB file or a FASTA sequence as input and predict the mutation is destabilizing or not on the basis of graph based approach and calculates ΔΔ G (http://structure.bioc.cam.ac.uk/mcsm) (Pires et al., 2013). For a range of proteins, mCSM provides better comparisons of mutations linked to disease. If the score is less than 0, the mutation reverses its effect.
DUET DUET (http://biosig.unimelb.edu.au/duet/) is a structure-based tool that takes a PDB file and an nsSNP mutation as input and calculates the DUET score, along with mCSM and SDM scores, as outputs. The DUET tool employs SVM and combines the results of mCSM and SDM to compute ΔΔ G. DUET integrates residue-level RSA (BY SDM) and secondary-structure and pharmacophore (by mCSM) features, and combines them with a supervised learning program (Pires et al., 2014).
MUpro MUpro (http://mupro.proteomics.ics.uci.edu/mutation_intro.html/) is a machine-learning technique used to determine how a single amino acid mutation affects protein stability. It employs two distinct machine-learning models to compute energy change. These programs are SVM and neural networks (Cheng et al., 2006). It computes a confidence score between -1 and 1 and the change in energy to quantify confidence. If the MUpro score is greater than 0, the mutation is predicted to increase protein stability; if the score is less than 0, it is predicted to decrease protein stability.
2.4. Identification of Pathogenic nsSNPs
Pmut PMut (http://mmb.irbbarcelona.org/PMut) is structure based tools which is used to check whether a non-synonymous mutation is related to disease or not (Lopez-Ferrando et al., 2017). Updated version of pmut determine. Pathogenicity of mutations, but it also provides an opportunity to train one's own prediction model on a particular dataset.
Phd-SNP Phd-SNP (http://gpcr.biocomp.unibo.it/cgi/predictors/PhD-SNP/PhD-SNP.cgi/) is a sequence or structure-based tool that is used to differentiate disease-related mutations regulated by the local environment of substitution. It takes a PDB file or a FASTA sequence as input.
RHAPSODY Rhapsody (http://rhapsody.csb.pitt.edu/) is a computational tool primarily used to determine the Pathogenicity of missense mutations based on sequence- and structure-based properties of the mutant protein (Ponzoni et al., 2020). It outperforms EV mutation and Polyphen-2 in analyzing residue-average pathogenicity.
2.5. Analysis of packing density and accessible surface area by using SDM2
The updated version of sdm2 also calculates the RSA, OSP, and residue depth for both mutant and wild-type proteins. For RSA, OSP, and residue-depth calculations, an environment-specific amino acid table is employed. RSA can calculate by the Lee and Richard technique. For the Structure stability analysis, OSP and residue depth are extended properties of the protein (DeDecker et al., 1996; Richards & Lim, 1993).
2.6. Analysis of noncovalent interactions
Arpeggio Arpeggio server is an structure based tool that takes a PDB ID or a PDB format file as input and estimates all the interatomic interactions of a protein. The Arpeggio server estimated mainly 15 different types of interatomic interactions, which can also be seen by this server (http://structure.bioc.cam.ac.uk/arpeggio/) (Jubb et al., 2017).
2.7. Analysis of aggregation propensity
SODA Solubility based on Disorder and Aggregation (SODA) (http://protein.bio.unipd.it/soda/) is a sequence or structure-based tool used to estimate the aggregation of protein, disorder, helix, and strand propensity that emerge because of mutations. Use the FASTA protein sequence or the PDB structure file as input. Insertion, deletion, substitution, and duplication (mutation type) were estimated by soda using PASTA 2.0, ESpritz-NMR, and Fells Soda score estimated according to the solubility difference between the mutant and WT of protein (Paladin et al., 2017).
3. Results and Discussion
3.1. Identification of Non-Synonymous Variants in TINF2
A list of 900 mutations was obtained from Ensembl, of which only 271 were non-synonymous. Out of 271 nsSNPs, only the structure of the N-terminal region (residue 2-202) is found in the PDB structure (PDB ID: 5XYF) of the TIN2 protein in the RCSB database. This analysis focuses on the sequence- and structure-based study of all nsSNPs. A multilevel approach was used to determine the functional and structural importance of the mutation on the TINF2 protein. To determine high-confidence deleterious and destabilizing non-synonymous mutations, sequence- and structure-based analyses were performed. All 271 non-synonymous mutations were subjected to sequence-based analysis using five tools: SIFT, PROVEAN, PolyPhen-2, Mutation Assessor, and SAAFEC-SEQ. Structure-based analysis is done by four different web-based tools, which are SDM2, DUET, mCSM, and MUpro, to estimate the 134 non-synonymous mutations present within the resolved crystal structure of the TINF2 protein (PDB ID 5XYF). For further studies, only high-confidence nsSNPs were included. Pathological analysis of high-confidence nsSNPs was performed using two web tools: PMut and PhD SNP.
3.2. Sequence-Based Prediction of Deleterious Mutations
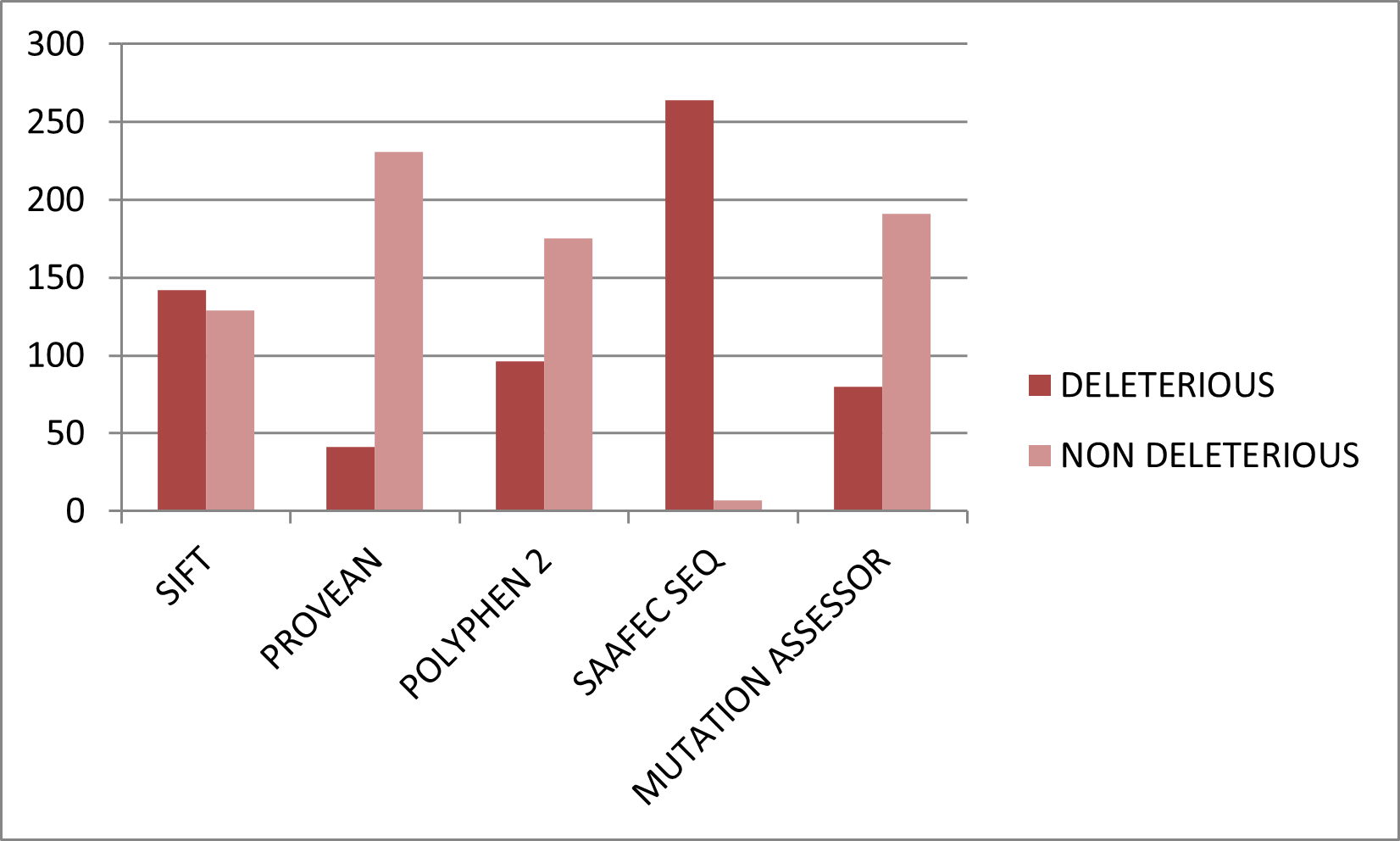
All 271 nsSNPs were analyzed using five independent sequence-based prediction tools. The proportion of variants predicted as deleterious varied among tools, reflecting differences in underlying algorithms. SIFT predicted 142 variants (52.3%) as damaging, PolyPhen-2 identified 96 (35.4%) as possibly or probably damaging, PROVEAN classified 41 (15.1%) as deleterious, Mutation Assessor detected 80 (29.5%) as functionally impactful, and SAAFEC-SEQ predicted 264 (96.3%) variants to reduce folding free energy (Table S1). Although individual tools yielded variable results, consensus analysis reduced the risk of false positives (Figure). Variants predicted as deleterious by at least four of the five tools were considered high-confidence functionally damaging mutations. This approach improved reliability by integrating evolutionary conservation, physicochemical changes, and predicted changes in folding energy. The high proportion of predicted deleterious variants suggests that many substitutions in TIN2 may disrupt its structural integrity or protein–protein interactions within the shelterin complex.
3.3. Structure-Based Stability Analysis
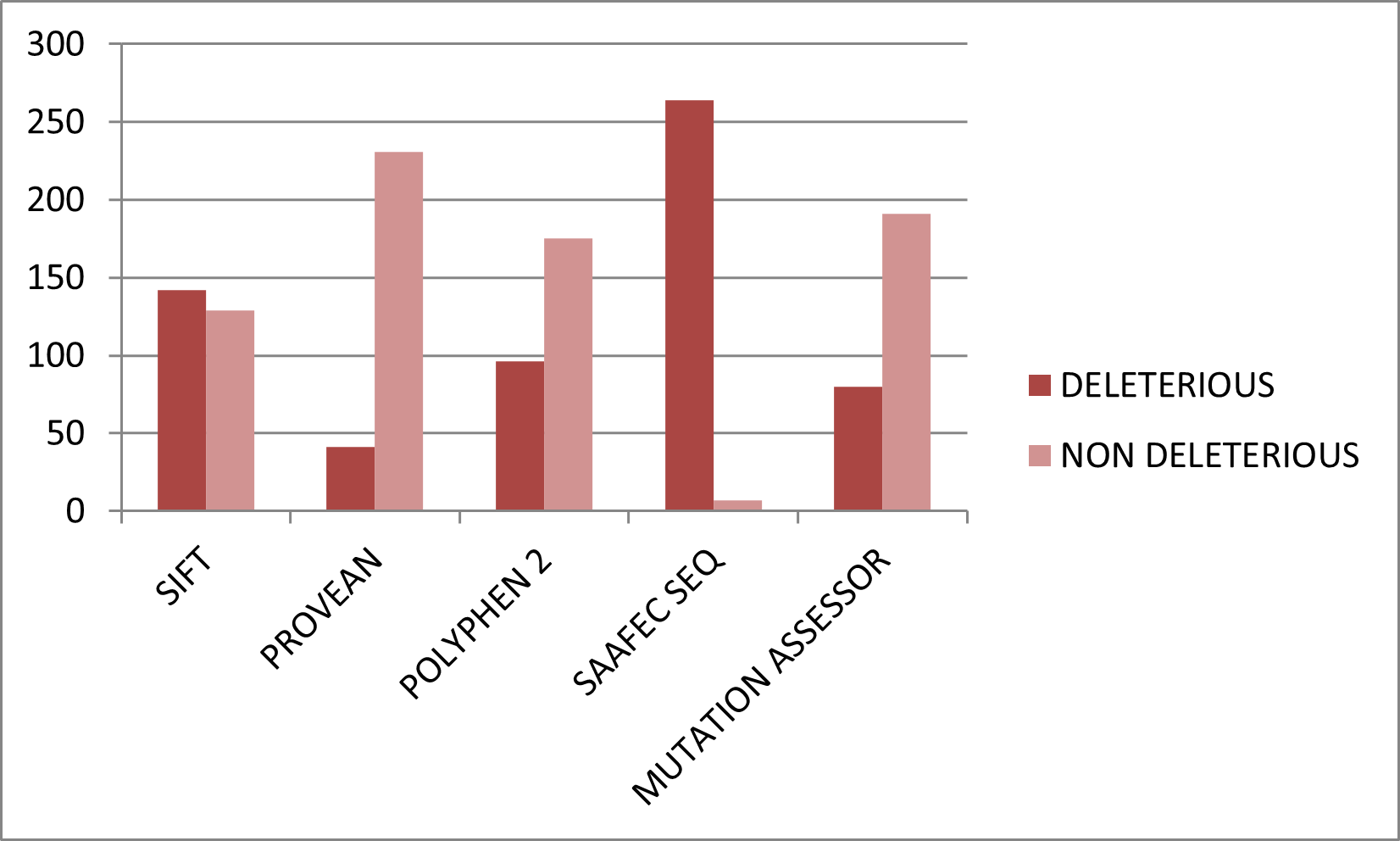
Structure-based prediction was performed on 134 nsSNPs located within the resolved N-terminal structure of TIN2. Stability changes were assessed using SDM2, DUET, mCSM, and MUpro. Among the analyzed variants, SDM2 predicted 58 (43.2%) as destabilizing, DUET identified 103 (76.8%), mCSM detected 120 (89.5%), and MUpro predicted 128 (95.5%) variants as decreasing protein stability (Figure). The predominance of destabilizing predictions indicates that many substitutions in the N-terminal domain may impair structural stability. Destabilization of TIN2 is particularly significant because the N-terminal region mediates high-affinity binding with TRF2 and contributes to scaffold formation within the shelterin complex. Structural perturbations in this domain may weaken intermolecular interactions, leading to defective telomere protection and compromised telomere length regulation. To increase prediction accuracy, only variants classified as destabilizing by at least three of four structure-based tools and as deleterious by at least four of five sequence-based tools were retained. This stringent filtering yielded 35 high-confidence deleterious and destabilizing nsSNPs.
3.4. Identification of pathogenic nsSNPs
The 35 high-confidence variants were further evaluated for disease association using PMut, PhD-SNP, and Rhapsody. PMut predicted 25 (71.4%) variants as pathogenic, PhD-SNP identified 24 (68.5%), and Rhapsody classified 11 (31.4%) as disease-associated. Twelve mutations, A16D, V22G, V34G, L38P, V49G, R52P, R56G, E109G, F114S, L121P, Y139C, and L150P, were consistently predicted as pathogenic by at least two of the three tools. These variants were selected for detailed structural investigation. Interestingly, several of these mutations are located in highly conserved residues within the N-terminal region, supporting their functional importance. Many involve substitution of small or hydrophobic residues with charged or bulky amino acids, which may significantly alter local structural packing.
Changes in Stability and Packing Density Analysis of relative solvent accessibility (RSA), occluded surface packing (OSP), and residue depth revealed that most of the selected mutations induced noticeable alterations in local packing density. Mutations such as V22G, V34G, V49G, and L150P replace hydrophobic residues with glycine or proline, thereby disrupting secondary-structure elements and reducing hydrophobic-core stability. Proline substitutions (L38P, L121P, L150P) are particularly disruptive due to conformational rigidity and their tendency to break α-helices. These changes likely perturb TIN2's folding and structural integrity (Table).
Alterations in Noncovalent Interactions Interatomic interaction analysis using Arpeggio demonstrated that several pathogenic mutations reduced hydrogen bonds, hydrophobic contacts, and ionic interactions compared to the wild-type structure (Table). Loss or rearrangement of these interactions can destabilize the local microenvironment and impair protein–protein interaction interfaces. For example, substitutions involving charged residues (R52P, R56G, E109G) may abolish electrostatic interactions critically for maintaining tertiary structure or mediating binding with TRF1 and TRF2.
Aggregation Propensity and Solubility Changes Aggregation analysis using SODA indicated that nine of the twelve selected mutations showed increased aggregation propensity, whereas the remaining three showed reduced solubility. Increased aggregation propensity suggests a higher likelihood of misfolding and potential formation of non-functional protein assemblies (Table). Protein misfolding or aggregation of TIN2 could disrupt shelterin complex assembly, thereby impairing telomere capping and exposing chromosome ends to DNA damage response pathways.
4. Conclusions
In this study, a comprehensive in silico framework was employed to systematically evaluate non-synonymous single-nucleotide polymorphisms in the TINF2 gene and to identify variants with potential pathogenic relevance in dyskeratosis congenita. By integrating multiple sequence-based, structure-based, and pathogenicity prediction tools, we prioritized high-confidence deleterious and destabilizing mutations affecting the TIN2 protein. Twelve missense variants (A16D, V22G, V34G, L38P, V49G, R52P, R56G, E109G, F114S, L121P, Y139C, and L150P) were consistently predicted to compromise protein stability and structural integrity. Detailed structural analyses indicated that these substitutions alter packing density, disrupt noncovalent interactions, and increase aggregation propensity, potentially impairing shelterin complex assembly and telomere protection. Given TIN2's central scaffolding role within the shelterin complex, these destabilizing effects may potentially contribute to telomere dysfunction associated with autosomal-dominant dyskeratosis congenita. Although the findings provide mechanistic insights into the structural consequences of TINF2 mutations, experimental validation is required to confirm their biological impact. The prioritized variants identified in this study may be suitable candidates for functional assays and genetic screening strategies. Overall, this work enhances understanding of the molecular basis of telomere-associated disorders and offers a systematic computational approach for investigating disease-related mutations.
Conflict of Interest
There is no conflict of interest to declare.
Data Availability Statement
The data supporting this study are provided in this article.
Funding
None
Acknowledgements
None
Supplementary Materials
None
Figures
Figure 1. Overview of the computational aspects to predict the pathogenic mutation in TINF2.
{kind=link}
Figure 2. Representation of the number of SNPs in TINF2 using the Ensembl database.
{kind=link}
Figure 3. Graphical representation of deleterious nsSNPs predicted by sequence-based tools.
{kind=link}
Figure 4. Graphical representation of deleterious and neutral nsSNPs predicted by structure-based tools.
{kind=link}
References
- 1
Sherry, S. T., Ward, M. H., Kholodov, M., et al. 2001, Nucleic Acids Research
- 2
Berman, H. M., Battistuz, T., Bhat, T. N., et al. 2002, Acta Crystallographica Section D
- 3
Cheng, J., Randall, A., Baldi, P. 2006, Proteins
- 4
Choi, Y., & Chan, A. P. 2015, Bioinformatics
- 5
DeDecker, B. S., O'Brien, R., Fleming, P. J., et al. 1996, Journal of Molecular Biology
- 6
Frescas, D., & de Lange, T. 2014, Journal of Biological Chemistry
- 7
Getov, I., Petukh, M., Alexov, E. 2016, Bioinformatics
- 8
Hubbard, T., Barker, D., Birney, E., et al. 2002, Nucleic Acids Research
- 9
Jubb, H. C., Ochoa-Montano, B., Pitt, W. R., et al. 2017, Journal of Molecular Biology
- 10
Kumar, P., Henikoff, S., Ng, P. C. 2009, Nature Protocols
- 11
Landrum, M. J., Lee, J. M., Riley, G. R., et al. 2014, Nucleic Acids Research
- 12
de Lange, T. 2005, Genes & Development
- 13
Lopez-Ferrando, V., Gazzo, A., de la Cruz, X., et al. 2017, Nucleic Acids Research
- 14
Paladin, L., Piovesan, D., Tosatto, S. C. E. 2017, Nucleic Acids Research
- 15
Pandurangan, A. P., Ochoa-Montano, B., Ascher, D. B., Blundell, T. L. 2017, Nucleic Acids Research
- 16
Pike, A. M., Strong, M. A., Ouyang, J. P. T., Greider, C. W. 2019, Molecular Cell
- 17
Pires, D. E. V., Ascher, D. B., Blundell, T. L. 2014, Nucleic Acids Research
- 18
Pires, D. E. V., Ascher, D. B., Blundell, T. L. 2013, Bioinformatics
- 19
Ponzoni, L., Penaherrera, D. A., Oltvai, Z. N., Bahar, I. 2020, Bioinformatics
- 20
Ramensky, V., Bork, P., Sunyaev, S. 2002, Nucleic Acids Research
- 21
Reva, B., Antipin, Y., Sander, C. 2011, Nucleic Acids Research
- 22
Richards, F. M., & Lim, W. A. 1993, Quarterly Reviews of Biophysics
- 23
Savage, S. A., Giri, N., Baerlocher, G. M., et al. 2008, Nature Genetics
- 24
Stenson, P. D., Mort, M., Ball, E. V., et al. 2009, Genome Medicine
- 25
Yang, D., He, Q., Kim, H., et al. 2011, Journal of Biological Chemistry
Tables
Table 1. Prediction of RSA, residue depth, and OSP for the wild-type and mutant TINF2 proteins using the SDM2 server.
| S. No. | Mutation | WT RSA (%) | WT Depth (Å) | WT OSP | MT RSA (%) | MT Depth (Å) | MT OSP | Outcome |
|---|---|---|---|---|---|---|---|---|
| 1 | A16D | 0.0 | 8.1 | 0.60 | 0.0 | 8.2 | 0.65 | Increased Solubility |
| 2 | V22G | 9.9 | 4.6 | 0.54 | 13.6 | 5.2 | 0.44 | Reduced Solubility |
| 3 | V34G | 0.4 | 7.5 | 0.53 | 17.0 | 7.0 | 0.47 | Increased Solubility |
| 4 | L38P | 0.3 | 8.6 | 0.52 | 6.2 | 8.1 | 0.51 | Increased Solubility |
| 5 | V49G | 7.1 | 5.1 | 0.42 | 34.4 | 4.1 | 0.26 | Increased Solubility |
| 6 | R52P | 37.7 | 3.9 | 0.31 | 13.3 | 4.8 | 0.42 | Reduced Solubility |
| 7 | R56G | 44.7 | 3.6 | 0.29 | 70.6 | 4.0 | 0.34 | Increased Solubility |
| 8 | E109G | 85.2 | 3.3 | 0.22 | 94.3 | 3.5 | 0.34 | Increased Solubility |
| 9 | F114S | 7.2 | 6.1 | 0.49 | 21.6 | 5.2 | 0.37 | Increased Solubility |
| 10 | L121P | 5.6 | 5.9 | 0.52 | 13.8 | 5.5 | 0.51 | Increased Solubility |
| 11 | Y139C | 12.9 | 4.8 | 0.49 | 9.1 | 5.3 | 0.45 | Reduced Solubility |
| 12 | L150P | 4.4 | 5.3 | 0.53 | 5.5 | 5.7 | 0.56 | Increased Solubility |
Table 2. Arpeggio server estimates of interatomic interactions for WT and mutant protein structures.
| S. No. | Variants | Vander Waals | Hydrogen | Ionic | Aromatics | Hydrophobic |
|---|---|---|---|---|---|---|
| 1 | Wild Type | 175 | 267 | 17 | 18 | 471 |
| 2 | A16D | 176 | 267 | 17 | 18 | 471 |
| 3 | V22G | 174 | 267 | 17 | 18 | 464 |
| 4 | V34G | 175 | 267 | 17 | 18 | 464 |
| 5 | L38P | 175 | 263 | 17 | 18 | 463 |
| 6 | V49G | 174 | 267 | 17 | 18 | 459 |
| 7 | R52P | 176 | 267 | 17 | 18 | 472 |
| 8 | R56G | 175 | 267 | 17 | 18 | 470 |
| 9 | E109G | 175 | 267 | 17 | 18 | 471 |
| 10 | F114S | 173 | 266 | 17 | 14 | 446 |
| 11 | L121P | 173 | 263 | 17 | 18 | 465 |
| 12 | Y139C | 172 | 268 | 17 | 13 | 451 |
| 13 | L150P | 175 | 268 | 17 | 18 | 472 |
Table 3. Prediction of aggregation propensity of mutant TINF2 protein using the SODA server.
| S. No. | Mutation | SODA | Remark |
|---|---|---|---|
| 1 | A16D | -2.9 | Less Soluble |
| 2 | V22G | 20.6 | More Soluble |
| 3 | V34G | 13.4 | More Soluble |
| 4 | L38P | 5.9 | More Soluble |
| 5 | V49G | 18.9 | More Soluble |
| 6 | R52P | 13.9 | More Soluble |
| 7 | R56G | 7.74 | More Soluble |
| 8 | E109G | 1.59 | More Soluble |
| 9 | F114S | 9.37 | More Soluble |
| 10 | L121P | 1.3 | More Soluble |
| 11 | Y139C | -6.5 | Less Soluble |
| 12 | L150P | -8.1 | Less Soluble |